Walk-through guide
Note: This program requires the Microsoft .NET framework version 2 (or later) to be installed on your computer.
Illuminator has been written specifically for targetted region- or gene-centred projects on the Illumina GAII sequencing platform (for example, highly parallel diagnostic mutation analysis). Many of its features reflect this goal, and are therefore not tailored to whole-genome (or transcriptome) projects. In this walk-through, the images will illustrate a step by step analysis of the p53 gene, in DNA from the HPAF pancreatic carcinoma cell line. The sample read data have been provided in the FASTA file named p53_Q-5.fasta. This was generated from the original raw data in the PRB format using the accessory program Illuminator data extractor (IDE), with a quality cut-off value of 5 (see the separate IDE guide for more information on this program). The sequence reads were aligned against the reference file p53.ref, which in turn was created by Illuminator from the genomic sequence in p53_gene.txt and the cDNA sequence in p53_cDNA.txt (as described below). All of these files are available via the Illuminator download page.
There are four main sections to this guide (matching the four tabs on the start-up window):
Information on the alignment algorithm used by Illuminator is contained in a separate Appendix.
Data analysis tab
The Data Analysis tab contains three panels. The Create reference file and Analysis panels allow the creation of reference files and analysis of data respectively, while the Progress panel is a visual guide to the progression of the data analysis (Figure 1).
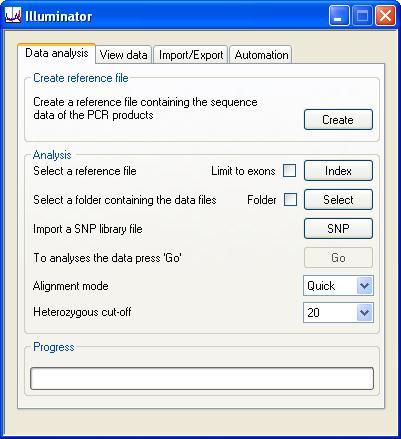
Figure 1
Creation of reference files
Reference files may be created using either Illuminator itself or the stand-alone Illuminator Reference Files program. Illuminator is better suited to creating a single reference file which will be used to analyse sequence data from a set of long PCR products, whereas Illuminator Reference Files is designed primarily for creating reference files for multiple genes in a specific genomic region. The creation of reference files using Illuminator is described below, while the user guide for Illuminator Reference Files is here and the program can be download from here.
To create a reference file, press the button on the Create reference file panel and select the text file containing the genomic reference sequence. Note: this sequence must contain the gene on the sense strand. Once selected, the Reference files window will be displayed (Figure 2).
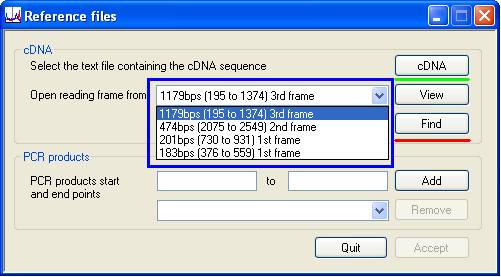
Figure 2
Next, press the button (above the green line in Figure 2) and select the plain text file containing the cDNA sequence. This sequence is screened for open reading frames (ORFs) which are then entered in the list-box (highlighted by the blue rectangle in Figure 2). The protein sequence of any ORF aligned to the cDNA sequence can be inspected by selecting it in the list box and pressing the button. To align the cDNA sequence to the genomic sequence, select the desired ORF and press the button (above the red line in Figure 2). The alignment will then be displayed in the Exon viewer window (Figure 3).
In this window, the upper line represents the cDNA sequence and the lower one the genomic sequence. The green rectangle indicates the position of the ORF, while the blue triangles map the locations of the exons from the cDNA sequence to the genomic sequence.
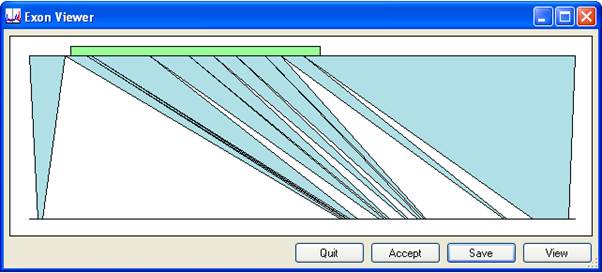
Figure 3
The Exon viewer window contains four buttons in the lower right corner, whose functions are:
- closes the window without accepting the alignment.
- closes the window and accepts the cDNA alignment.
- saves the alignment as a plain text document.
- displays the alignment as a text file.
If the alignment is accepted, the button at the lower right of the Reference files window will become enabled and pressing it will generate the reference file (Figure 4). However, the resultant reference file will include all the sequence in the genomic sequence file. When analysing data derived from long PCR products, or in order to disregard non-coding sequences, it may be desirable to restrict the sequence included in the reference file to regions of interest. Doing this will both decrease the size of the reference file and speed up the analysis.
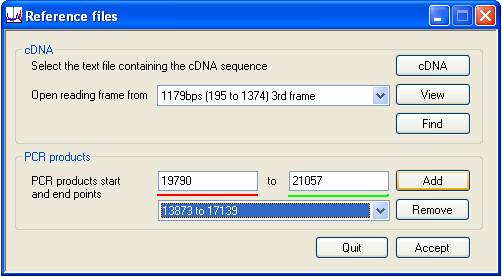
Figure 4
To select such a region of interest, enter its start and end points into the two text boxes above the red and green lines and press the button to their right. This clears the text boxes and adds the chosen region to the list box below the red and green lines. (To remove a region, select it from the list box and press the button). Finally, when all of the desired regions have been entered, press the button at the bottom right of the window and enter the name of the reference file.
Aligning read data to a reference file
To simplify the alignment process, only two user-defined options can be set: the “Alignment mode”, (Figure 5, underlined in black) which is described in the Appendix page, and the “Heterozygous cut-off” percentage (Figure 5, underlined in red). A minor sequence variant must exceed this frequency for the position to be considered heterozygous. The analysis work-flow is described below:
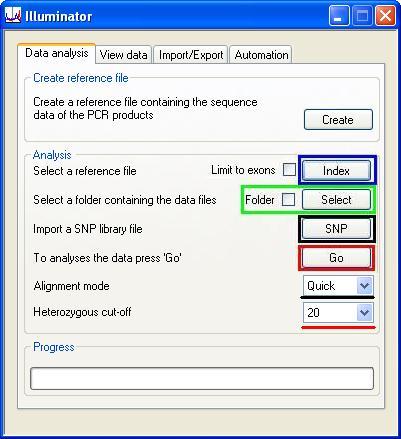
Figure 5
- Press the button (Figure 5, blue rectangle) and select the reference file. The program reads the reference file and indexes the sequence (as outlined in the Appendix).
- If you are screening a batch of sequence text files (*_seq.txt) in a directory, tick the box next to the button (Figure 5, green rectangle). If you are analysing a single FASTA file, leave this box unticked. Accordingly, press the button and select either a single file (box unticked) or a folder of files (box ticked; n.b. if you select a folder, Illuminator will try to read all text files, so do not put text files other than sequence reads in the correct format in this folder).
- If you have a file containing SNP data (*.sref) extracted from a previous analysis, press the button (Figure 5, black rectangle) to load this file. (Making SNP data files is described in the SNP Library section)
- Select the alignment mode from the list-box (above the black line in Figure 5). Each of these options is explained at the end of the Appendix page.
- Select the “heterozygous cut-off” using the list-box at the bottom of the interface (above the red line in Figure 5). If this value is set at 20, then each allele must comprise at least 20% of the total reads to be called heterozygous.
- Finally press (highlighted by the red rectangle in Figure 5) to align the sequence reads against the reference sequence. Illuminator first identifies SNPs in the patient sequence data and adds them to the reference sequence. The program then reanalyses the read data, mapping reads containing multiple sequence variants as well as identifying the positions of indels. While it does this, it creates a *.temp file which contains reads that it thinks span the indels. Finally, it maps the reads in the *.temp file to the reference sequence containing possible indel. When this step has been completed, the *.temp file is deleted. Since the file is linked to the current analysis, data in one folder cannot be analysed simultaneously by two instances of Illuminator. As the analysis is performed, its progression is shown as a series of coloured bars in the Progress panel. The progress bars fill the meter after analysis of 1 million sequence reads, after which it resets to zero and starts again.
- Once the alignment is completed, the interface automatically displays the View data tab, containing the Display options panel.
View data tab
The View data tab (Figure 6) contains four buttons, which invoke windows that respectively display the complexity of the reference sequence, a global view of the alignment, a local view of the alignment read depth, and information related to each indel that the analysis has found.
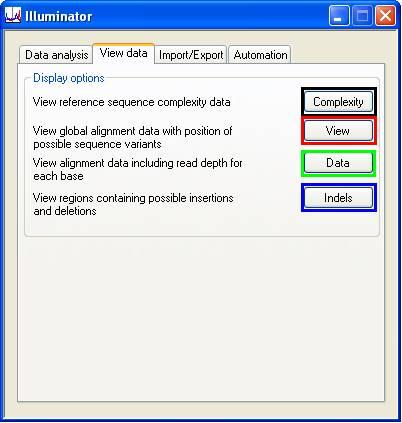
Figure 6
General features of the ‘Complexity’, ‘View’ and ‘Data’ view forms
The first three buttons all generate windows that display their data pictorially. Mouse-clicking on any image selects that point as the active position in the reference file. If two or three windows are open, redefining the active point within one will also change it in the others, so that each window displays information about the same point. Similarly, each window has a list box in its lower panel; this selects data related to each region of interest (e.g. a long PCR product). Again, changing this in one window also changes it in the other open windows.
The PCR products are all equally scaled, so that the longest one spans the image. Exonic data is shown as a pale blue rectangle and protein-coding sequences in pink (for example, see Figure 7). These windows can be resized.
The fourth button generates a window that behaves differently.
Complexity view
Pressing the button (Figure 7, black rectangle) opens the Complexity view window, comprising two images. The upper image, in the Reference sequence statistics panel, shows a global view of the "uniqueness" of the sequence across a region of interest. The lower Sequence panel displays an image of the local data complexity, along with the DNA sequence itself.
The blue histogram in the upper image (black arrow in Figure 7) charts how commonly the octamer at that position occurs in the reference sequence (all PCR products). Regions that contain runs of common octamers may be susceptible to incorrect alignments.
Clicking on the image selects that point as the active sequence position; this is identified by the vertical red line in the upper image and by two vertical red lines in the lower image; the octamer at that position is highlighted as a pale blue rectangle (outlined in black for emphasis in Figure 7). The vertical numbers in the lower panel are counts of the frequency with which the octamer at that position occurs in the entire reference sequence. In the example shown, the selected octamer occurs 4 times, while the octamers to its right occur at a total of 22 positions. If any of these octamers occur elsewhere in the same region of interest, their positions are flagged by short vertical green lines. (One is highlighted in the green rectangle in Figure 7.) If such an octamer is in the opposite orientation, its green line is displaced to a lower position than if it is in the same orientation.
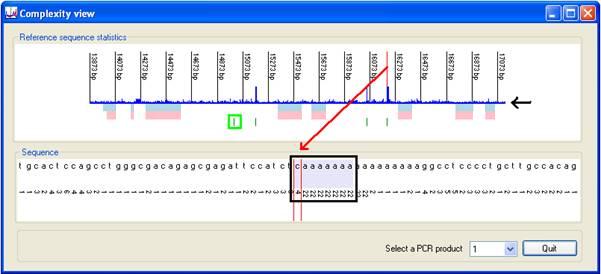
Figure 7
Global view window
Pressing the button (red rectangle in Figure 6) displays the Global view window, which gives a global view of the alignment information (Figures 8 and 9). This window shows the positions of single base variants and indels in the sequence. As with the Complexity view window, clicking on either of its two panels selects that position on the image as active, and centres the lower image on the active nucleotide (indicated by red vertical lines). The upper part of the display, in the Alignment statistics panel, includes line plots of the read depth across the region of interest, blue for sequences originating from the sense strand and red for antisense. Occasionally, a large spike may occur in the graph due to the presence of a sequence that is present at very high copy. The presence of such spikes may reduce the scaling of the Y axis, flattening the graph and obscuring the read depth data. To overcome this, the Y axis may be scaled logarithmically by ticking the box in the bottom left hand corner of the window.
Below the pink and blue rectangles representing the exonic sequences are five rows that highlight the locations of SNPs and indels. The uppermost of these displays a red vertical line at the position of single base variants. To be placed in this row, a variant must be identified by different sequence reads mapping to both the forward and reverse complement of the reference sequence. If a variant is identified only by reads mapping to the forward sequence (or only to the reverse complement) its position is marked by a blue vertical line in the second (or third) rows. Similarly, indels are annotated by red lines in the fourth row, only if identified in different sequences aligned both to the forward and reverse complement reference sequence. If not found in both forward and reverse sequence reads, an indel's position is shown in the fifth row. When a patient is heterozygous for an indel, it may be associated with a number of incorrectly called sequence variants. Such spurious calls will be shown in this window, but will be ignored (under default settings) when exporting variant data to a text file.
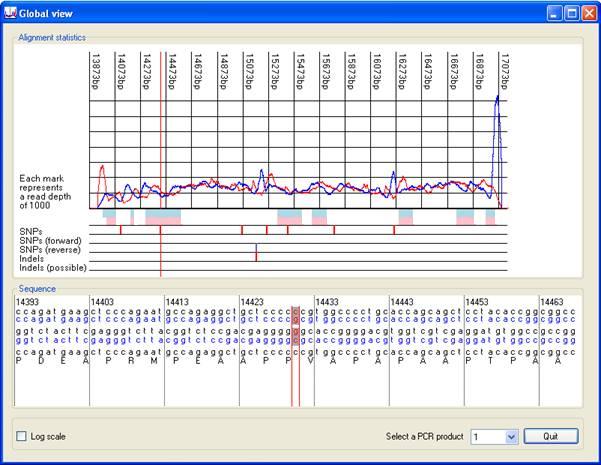
Figure 8
The lower Sequence panel shows the local sequence flanking the selected position. This appears as up to six lines of sequence;the first line (black text) is the sense strand of the reference sequence, the second (blue text) is the sequence deduced from the experimental data, in which heterozygous changes are shown as y, r, w, s, m or k. (The program does not tolerate three different nucleotides at one position). Similarly, the next two lines show the antisense reference and deduced (patient) sequences. The last two lines contain the exonic and protein sequences derived from the reference file (not the patient data). If the active position is in an intron, these lines will be absent. Single-base variants visible in the lower panel are identified as red text on a grey background. For example, in Figure 8, the active position contains a homozygous C→G substitution.
If the selected position is close to an indel, two further lines of text will be displayed, showing the deduced patient sequences across the indel, from reads matching the sense strand (upper line) and antisense strand (lower line). Figure 9 shows an example of a 3-nt deletion (g.15188_15190delAAA) within a run of 18 A residues.
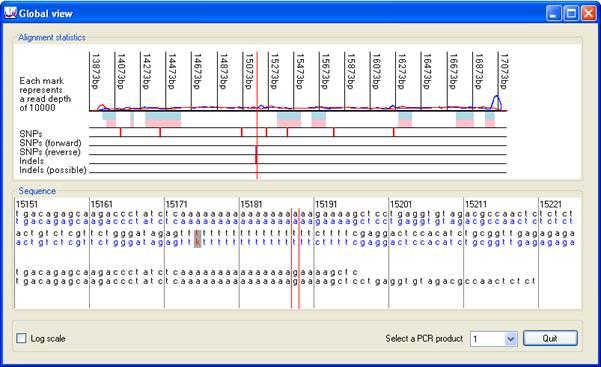
Figure 9
Data view window
Pressing the button (green rectangle in Figure 7) opens the Data view window. This only displays local sequence data around the selected site. While it is possible to navigate along the sequence using the scroll bar below the image or by clicking on the image, it is usually easier to move to a specific position by selecting that point in either the Global view or Complexity view window.
The alignment data for sequence reads mapped to the sense strand is shown in the upper half of the panel (labelled A-E in Figure 10) and the data for reads aligned to the anti-sense strand is displayed in the lower half (a-e in Figure 10). As in the Global view window, substitutions are highlighted with a grey background and the current position is shown by two vertical red lines (Figure 10). This window shows the deduced (patient) and reference sequences, along with read depth information as outlined below:
- A and a: The deduced (patient) sequence heterozygous positions are identified by the use of the Y, R, W, S, M or K IUPAC ambiguity symbols.
- B and b: Graphical representation of the relative proportion of each nucleotide mapped at a specific position. The image imitates an ABI-style electropherogram, in that it is composed of four coloured lines (A = green, C = blue, G = black, T = red). If 100% of the reads identify the same base, the peak will touch the black dashed line, whereas if a position is heterozygous and one nucleotide is present in ~50% of the reads, its peak will reach about half way to the dashed line from the baseline. (n.b. Figure 10 shows data derived from a different cell line from that used in the data supplied for download.)
- C and c: The reference genomic sequence is shown below the graphic. If the displayed region includes any exonic or protein sequence, extra lines are written to display this (below the line labelled C in Figure 10), for the forward (sense) direction of the reference sequence only.
- D and d: This line is the result of deconvoluting the deduced (patient) DNA sequence to identify sequence changes. Where the patient and reference sequence are the same, this line will be identical to the reference sequence. Where the patient and reference sequences differ, this line displays the patient sequence if it is homozygous. However, where a position is heterozygous, Illuminator subtracts the reference sequence from the patient sequence, to identity the variant nucleotide. For example, if the reference and patient sequences are ACGCGT and ACSGGT, respectively, this line will display the novel sequence ACCGGT.
- E and e: This line shows the read depth at each position.
- F: The position in the original genomic reference sequence used to create the reference file is annotated, above the position in the long PCR product (which is shown in brackets).
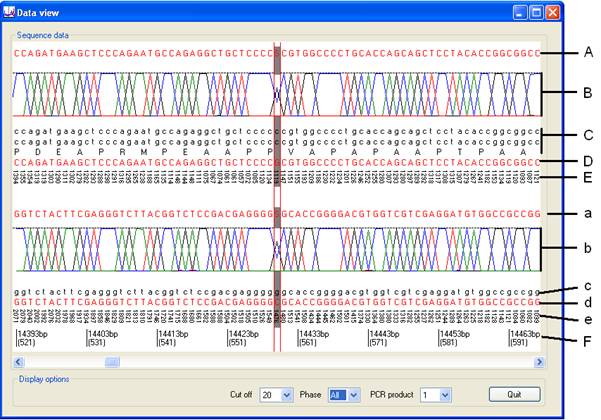
Figure 10
Indels window
This window works differently to the other three, being purely text-based. Each possible indel is viewed in turn, by selecting it from one of the list boxes in the View an Indel panel at the bottom left. The first list box, labelled ‘All’, lists all the positions Illuminator has identified as containing a possible indel. The list labelled ‘True’ contains the positions Illuminator has identified as true indels. When analysing for indels, the sense and antisense strands are processed independently of each other, and an indel is called only if each strand gives the same answer. This methodology is reflected in the Indels view interface, which shows the forward strand analysis (labelled A-E in Figure 11) in the upper half of the interface and the reverse strand analysis (labelled a-e) separately below it.
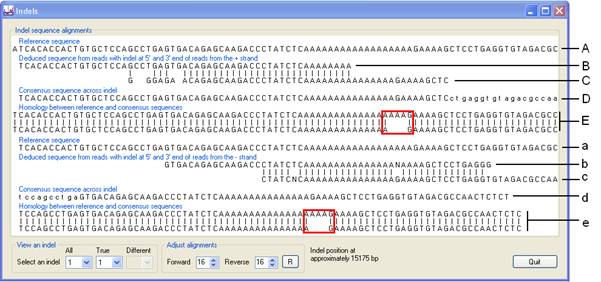
Figure 11
The first line of each analysis (A and a in Figure 11) is the reference sequence spanning the putative indel. Below these are the results of the sequences aligned to the strand 5’ to the indel (B and b in Figure 11) and the result of reads aligned to the strand 3’ to the indel (C and c in Figure 11). Lines D and d show the patient’s consensus sequence across the indel derived by aligning the sequences B to C and b to c. Finally, the alignment of the reference sequence and the patient consensus sequence is shown (E and e in Figure 11). In Figure 11 the alignment identifies the mutation to be a 3-nt deletion (red box). Only if the same indel is found in both E and e alignments is the indel annotated when the sequence variants are exported; otherwise, the estimated approximate position of the indel will be listed.
The consensus sequences (D and d) can be manually adjusted using the input boxes in the Adjust alignments panel. Changing the number in the ‘Forward’ list-box moves the position of the sequence in line C relative to the B line sequence, which in turn changes the consensus sequence in line D and the alignment E. Similarly, editing the value in ‘Reverse’ list-box affects the sequences in lines c, d and e. Pressing the button resets these values to the original offset. If these values are changed, the new alignment will be used when annotating and exporting the sequence variants.
Import/Export tab
The Import/Export tab contains three panels (Figure 12).
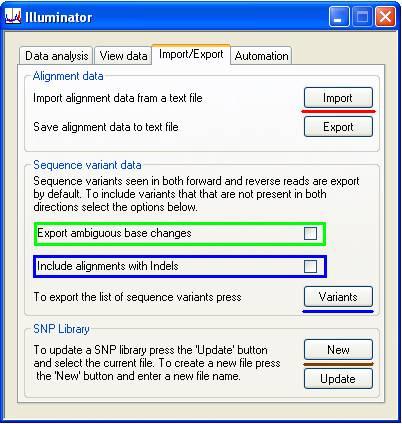
Figure 12
Alignment data
This panel contains two buttons; (below the red line in Figure 11), which saves the analysis data to a file, and , which opens a previously saved data analysis. There is no difference between re-analysing a run and opening the same analysis saved to a file; however, the exported data file is much smaller than the data file containing the sequence reads. This option therefore makes it possible to e-mail analysis data to co-workers, etc.
Sequence variant data
Pressing the button on the Sequence variant data panel exports the sequence variants to a plain text file (Table 1). If a variant occurs in the open reading frame, the variant codon is shown together with the equivalent codon in the genomic and cDNA sequences. The position of the mutation in the cDNA sequence is specified relative to the first nucleotide of the start codon. If a sequence variant lies within a splice site, this is indicated in the third column (which normally contains the protein sequence data). Currently, the output does not state whether an indel is heterozygous or it's read depth.
| Mutation in genomic sequence | Mutation in cDNA | Mutation in protein sequence or splice site | Homozygous or heterozygous | Read depth |
|---|---|---|---|---|
| g.14117C>G | intronic | non-coding | (homozygous) | (a: 0, c: 1, g: 1150, t: 0) |
| g.14430C>G | intronic | non-coding | (homozygous) | (a: 3, c: 15, g: 1678, t: 2) |
| g.15065T>C | intronic | non-coding | (homozygous) | (a: 3, c: 2715, g: 0, t: 0) |
| g.15188_15190delAAA | intronic | non-coding | ||
| g.15257G>A | intronic | non-coding | (homozygous) | (a: 3238, c: 14, g: 9, t: 2) |
| g.15423C>T | c.451C>T | p.151P>S (Gene: dCCC-P, cDNA: dCCC-P, Variant:dTCC-S) | (homozygous) | (a: 0, c: 7, g: 1, t: 2043) |
| g.15787A>G | intronic | non-coding | (homozygous) | (a: 5, c: 0, g: 2486, t: 0) |
| g.16258G>C | intronic | non-coding | (homozygous) | (a: 2, c: 2343, g: 5, t: 2) |
Table 1
Selecting the ‘Export ambiguous base changes’ option (green rectangle in Figure 12) exports variants that appeared in only one strand (e.g. that appeared in reads aligned to the sense strand but not the antisense strand). It also exports single-base variants close to suspected indels (which are probably artefacts caused by the indel).
Selecting the ‘Include alignments with Indels’ option (blue rectangle in Figure 12) exports the alignment of the consensus and reference sequences with the annotated indel. These alignments are analogous to the alignments labelled E and e in Figure 11.
SNP Library
With each successive analysis of a gene, the positions of an increasing number of SNPs and single-nucleotide mutations will be identified. These variants can be exported, via the SNP library panel. To start a new data set, press the button (above the brown line in Figure 12) and select a file name. In subsequent analyses of the same gene (using the same reference file), the library can be updated by pressing the button (below the brown line in Figure 12) and selecting the library file. The variant data is updated and then exported to a new file with the same file name as the original, except that the current data is appended to it. This file is saved as a plain tab-delimited text file, which can be opened and edited using any text editor or spreadsheet program. (If opened in Excel, the file must be saved as a tab-delimited file and not as a worksheet.) If this file is loaded before analysing sequence data using the button on the Analysis tab, the positions of the sequence variants are shown (in the Global view window) as vertical lines over the blue and pink rectangles representing the exonic sequences (Figure 13).
Initially, each sequence variant is classified as 'Untyped' in the file and is displayed as an orange line. However, once the significance of the variant is known, it can be classified as either 'Polymorphism' or 'Mutation'. The variant will then be indicated by a blue or red line, respectively. Table 2 shows a partially edited SNP file, where the first two variants are untyped and the remaining variants classed as 'Mutation' or 'Polymorphism'. When this file is used together with the p53 sequence data, the positions and classes of the variants are visualized relative to those of the variants found in the sample data. The exact nature of each known sequence variant is also highlighted in the lower panel (Figure 13). In Figure 13, the positions of p53 mutations found in a number of cell lines, including one variant which is identified in the current sequence data, can be seen.
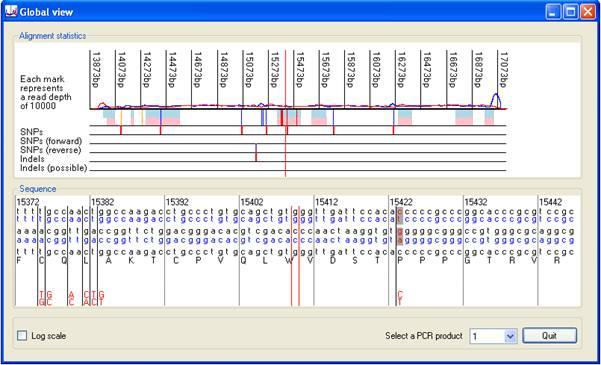
Figure 13
The first line of a ‘SNP’ file contains the name of the reference file used to analyse the data. Only data analysed using this reference file can be used to view or update the information in a SNP file. The final column of the file shows the number of individuals that mutation has been found in (not the number of chromosomes it has been found on).
| Variant | cDNA | Protein | Class | Occasions seen |
|---|---|---|---|---|
| p53.ref | ||||
| g.14117C>G | intronic | non-coding | untyped | 2 |
| g.14283C>A | intronic | non-coding | untyped | 2 |
| g.14430C> | c.215C>G | p.73P>R (Gene: ccc-P, cDNA: ccc-P, Variant:cgc-R) | Polymorphism | 2 |
| g.15065T>C | intronic | non-coding | Polymorphism | 4 |
| g.15223T>C | intronic | non-coding | Polymorphism | 2 |
| g.15231G>A | intronic | non-coding | Polymorphism | 2 |
| g.15257G>A | intronic | non-coding | Polymorphism | 4 |
| g.15375T>G | c.403T>G | p.135C>G (Gene: tgc-C, cDNA: tgc-C, Variant:ggc-G) | Mutation | 1 |
| g.15376G>C | c.404G>C | p.136C>S (Gene: tgc-C, cDNA: tgc-C, Variant:tcc-S) | Mutation | 1 |
| g.15379A>C | c.407A>C | p.137Q>P (Gene: caa-Q, cDNA: caa-Q, Variant:cca-P) | Mutation | 1 |
| g.15381C>A | c.409C>A | p.137L>M (Gene: ctg-L, cDNA: ctg-L, Variant:atg-M) | Mutation | 1 |
| g.15382T>C | c.410T>C | p.138L>P (Gene: ctg-L, cDNA: ctg-L, Variant:ccg-P) | Mutation | 1 |
| g.15383G>T | c.411G>T | p.138L>L (Gene: ctg-L, cDNA: ctg-L, Variant:ctt-L) | Mutation | 1 |
| g.15423C>T | c.451C>T | p.151P>S (Gene: ccc-P, cDNA: ccc-P, Variant:tcc-S) | Mutation | 1 |
| g.15496G>A | c.524G>A | p.176R>H (Gene: cgc-R, cDNA: cgc-R, Variant:cac-H) | Mutation | 1 |
| g.15787A>G | intronic | non-coding | Polymorphism | 4 |
| g.16258G>C | intronic | non-coding | Polymorphism | 1 |
| g.20673G>A | intronic | non-coding | Polymorphism | 2 |
Table 2
Automated analysis
When aligning a number of FASTA sequence files, originating from different samples, against one or more reference files, it is possible to automate the analysis using the Automation tab (Figure 14). To do this, first select the folder that contains the reference file(s) to be used, by pressing the button. Next, press the button to select the folder of FASTA files containing the short read sequences. Finally, pressing starts the automated analysis. For each FASTA file / reference file combination, Illuminator will save the alignment data and a list of the variants identified, in the same folder as the FASTA files. The saved alignment can be re-entered into Illuminator using the button on the Import/Export tab, as described above. The alignment file is named as follows: "FASTA file name" + "_" + "reference file name" + ".dref", while the sequence variants are saved in the "FASTA file name" + "_" + "reference file name" + "_Mutations.txt" file. If the box is ticked, the analysis ignores all reads aligned to intronic sequences more than 50 bp from an exon.
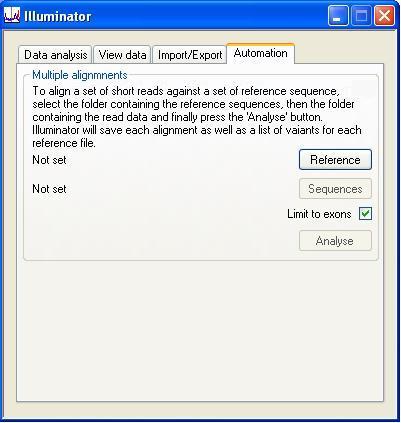
Figure 14