Illuminator alignment algorithm
General considerations
Illuminator was designed with the aim of screening individual genes for mutations, rather than screening large regions containing many genes. A number of practical considerations therefore influenced the program design. For example, in a hunt for an unknown disease-causing mutation, there is often in practice no way to prove that rare intronic sequence variants are disease-causing. Therefore, the program has a facility to create reference sequences that only include exons (along with limited flanking intronic sequences). The relatively small size of such reference sequences makes it possible to quickly align Illumina reads using a mismatch-tolerant algorithm. Also, due to the comparatively small region to be aligned, it becomes fairly straightforward to screen a number of genes by pooling several long PCR products in a single GA-II lane and then screening each gene in turn.
Indexing the reference sequence
Central to the alignment algorithm is an index of all 65536 possible 8-nt DNA sequences (octamers). The position of each successive octamer within the reference sequence is linked to the entry for the corresponding octamer sequence in this index. This means that any 8-nt test sequence can be rapidly looked up in the index, defining its location(s) in the reference file (Figure 1). Some octamers will not occur in the reference sequence, while others will be relatively abundant. Since the sequence reads originate from both the forward and reverse complement of the reference DNA sequence, the program automatically reverse-complements the genomic reference sequence and appends this to the forward sequence. This obviates the need to reverse-complement each experimental read and search for homology twice.
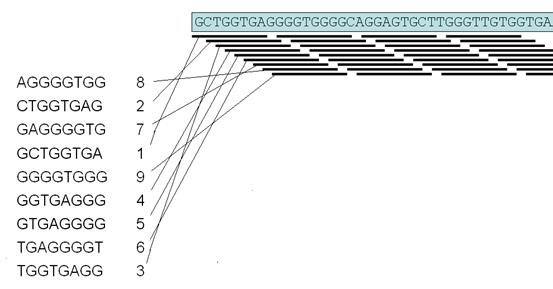
Figure 1
Mapping sequence reads against the reference file
The mapping process includes two cycles of data alignment. In the first cycle, information is collected on the locations of SNPs, which is then used to update the reference index. The data is then read again to allow the detection of closely spaced SNPs during the second cycle. After this, the locations of indels are found and mapped, using a subset of reads collected during the second cycle. These steps are performed as follows:
First cycle
Each read is broken down into 4 non-overlapping octamers and the octamer positions are found in the reference sequence by consulting the index of octamers. The program then searches for a set of positions whose locations in the reference file increase stepwise by 8 nt for each successive octamer (Figure 2). The sequence read is then mapped to the reference sequence, to identify the positions of any sequence variants within the read that lie outside the region containing the four octamers.
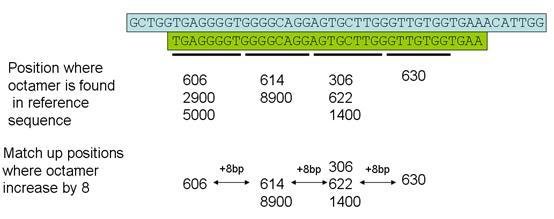
Figure 2
If the read does not contain a run of 4 sequential matching octamers as described above, Illuminator next tries to identify sets of octamers where the first and fourth octamers have the expected separation, along with one or other of the internal octamers (Figure 3). Again, the sequence read is then mapped to the reference sequence to identify the positions of any sequence variants.
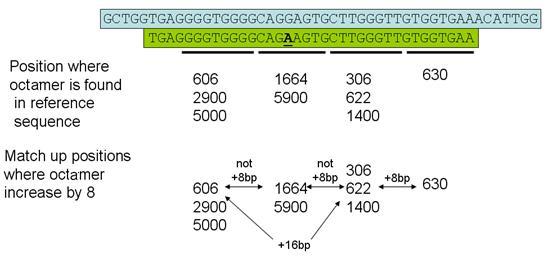
Figure 3
Finally, any read which has not yet been mapped is checked for sets of octamers that include 3 consecutive positions in the reference sequence, as shown in Figures 4 & 5. As before, these reads are mapped on to the reference sequence and the position of any sequence variants are noted.
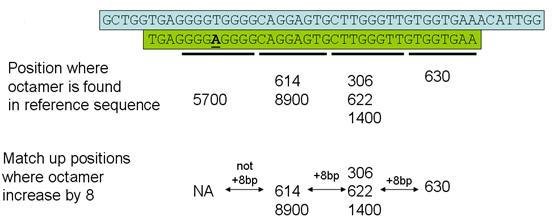
Figure 4
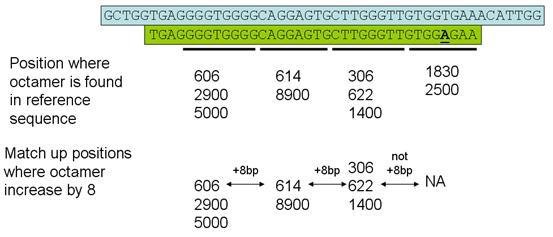
Figure 5
Second cycle
All the sequence variants found in the first cycle are now superimposed on the reference sequence, which is then re-indexed to include both the original reference sequence and the sequence variants found in the first alignment cycle. By re-indexing the reference sequence in this way, it is possible to map reads which contain multiple genuine sequence variants (such as a mutation close to a SNP). Such reads would have failed to be aligned in the first cycle, because only two octamers would have been mapped close to each other.
The alignment process is now re-run as before, except that this time any read that is found to be mismatched at either end of the sequence (i.e. Figure 4 or 5) is stored to a temporary file. These reads are important since they are the only class of read containing insertions or deletions that can easily be mapped. Once the second round of analysis is complete, the locations of all the sequence variants are noted. This information is then used to identify single nucleotide variants, including both SNPs and potential pathogenic point mutations. Also, the read depth is calculated from this alignment data, with the data split into three "phases"; the first phase comprises all the reads aligned as shown in Figures 1 and 2, the second phase comprises reads aligned with a 5′ mismatch (Figure 4), and the third phase consists of reads with a 3′ mismatch (Figure 5). This information can all be inspected within the Data view window, by selecting the "Phase" value using its list-box. (See Figure 10 in the Illuminator guide).
Mapping indels
Through empirical observations of alignment data, we found that short regions containing a large number of variants are strongly suggestive of the presence of an insertion or deletion. Therefore, Illuminator checks the positions of variants to identify regions where an unusual number of substitutions occur together within a region of 16 nt. (These variants are required to be present both in reads mapped to the forward strand and in reads mapped to the reverse complement strand of the reference sequence.) The pattern of a 3-nt deletion in a poly(A) stretch can be seen in Figures 6 (5′-mismatched sequences) and 7 (3′-mismatched sequences) which are screen-shots from the Data view window of Illuminator.
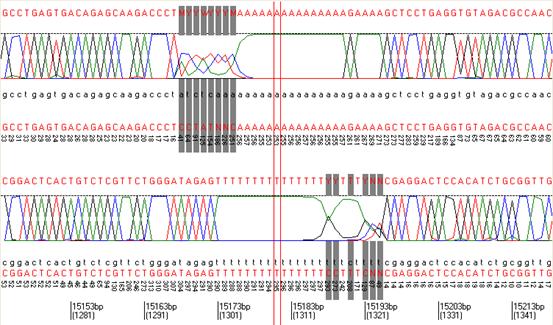
Figure 6
The location of the possible indel is noted and the flanking sequence is aligned to the sequence reads placed in the temporary file during the second cycle of read alignment. This alignment is done in the same manner as shown in Figures 4 and 5, but the alignments for the 5′ and 3′ mismatches to the forward and reverse-complement sequences are kept separate, and used to create four sequences of about 50 nt in length. The 5′- and 3′-mismatched sequences are then aligned to create a consensus sequence across the indel (for both the forward and reverse-complement sequences). These consensus sequences are then aligned to the reference sequence, in order to identify the location and sequence of an indel. Only indels that are independently identified on the forward and reverse-complement sequences are annotated and exported. The sequence of these alignments can be viewed in the Indels window of Illuminator, as described in the Indels window section of the Illuminator guide.
Alignment mode
When mapping reads, Illuminator searches for positions with at least 3 out of 4 correctly placed octamers, as described above. Consequently, a read may be mapped to locations where (A) all four octamers are aligned (Figure 2), (B) the first and last octamer plus either the second or third are aligned (Figure 3), or (C) three consecutive octamers are aligned (Figures 4 and 5). Illuminator ranks these alignments as A > B > C, so that if a read is matched to multiple locations, alignments meeting criterion A are scored higher than criterion B, which in turn ranks higher than C.
Illuminator has four different modes of alignment, , , and . These options control how short reads, which can be mapped to more than one position, are placed within the alignment created by the second cycle, as follows:
- This option places a short read at the first location that matches any of the three different criteria. Once this position has been identified, Illuminator makes no further attempt to find other locations to which the short read could also be mapped.
- This option finds all the possible locations where a short read could be matched. If a read can be matched to a number of sites, it is finally mapped to the first location of the highest scoring criterion, i.e., if a read is mapped to positions 100 bp and 200 bp by criterion A and 50 bp by criterion B, the read will be mapped to position 100 bp.
- If a read can be mapped to a number of locations, it is mapped to all the location with the highest score. For example, if a read is mapped to positions 100 bp and 200 bp by criterion A and 50 bp by criterion B, the read will be mapped to both 100 bp and 200 bp but not to 50 bp.
- If a read can be mapped to multiple locations, it is placed randomly at a location which meets the highest-scoring criterion. For example, when a read can be placed at positions 100 bp and 200 bp by criterion A and 50 bp by criterion B, the read will be randomly assigned to position 100 bp or 200 bp.
To illustrate the different effects of each alignment mode, Table 1 shows the position a read is assigned to, when each of the three criteria map the sequence to different places in the reference sequence.
| Possible locations using: | Position read aligned too using option: | |||||
|---|---|---|---|---|---|---|
| Criterion A | Criterion B | Criterion C | Quick | First | All | Random |
| 100 | 200 | 300 | 100 | 100 | 100 | 100 |
| 200 | 300 | 100 | 100 | 200 | 200 | 200 |
| 300 | 100 | 200 | 100 | 300 | 300 | 300 |
| 100 and 400 | 200 and 500 | 300 and 600 | 100 | 100 | 100 and 400 | 100 or 400 |
| 200 and 500 | 300 and 600 | 100 and 400 | 100 | 200 | 200 and 500 | 200 or 500 |
| 300 and 600 | 100 and 400 | 200 and 500 | 100 | 300 | 300 and 600 | 300 or 600 |
| - | 200 | 300 | 200 | 200 | 200 | 200 |
| - | 300 | 100 | 100 | 300 | 300 | 300 |
| - | 200 and 500 | 300 and 600 | 200 | 200 | 200 and 500 | 200 or 500 |
| - | 300 and 600 | 100 and 400 | 100 | 300 | 300 and 600 | 300 or 600 |
| - | 100 and 400 | 200 and 500 | 100 | 100 | 100 and 400 | 100 or 400 |
| - | - | 200 | 200 | 200 | 200 | 200 |
| - | - | 200 and 500 | 200 | 200 | 200 and 500 | 200 or 500 |
Table 1