Walk-through guide
Note: This program requires the Microsoft .NET framework version 2 (or later) to be installed on your computer.
The Illuminator_data_extractor program was designed to convert various output data file formats generated by an Illumina clonal sequencer to a format that Illuminator can process. While Illuminator itself can analyse files in the fasta, _seq.txt and _qseq.txt file formats, it was decided for logistical reasons to separate the task of data analysis from the task of file formatting. Therefore this data extraction program will be updated in response to changes in Illumina output file formats, while Illuminator will be updated in response to other changes such as increased read length.
It was also decided to concentrate on the formats used by sequence files that have not been subjected to quality scoring by comparison of the reads to a reference sequence. This is because some of the capabilities for which Illuminator was designed (such as detection of rare sequence variants, and the processing of pooled samples identified by 5’-end tags) would be adversely affected by such comparison. For example, reads containing genuine sequence variants (or 5' tags) might be rejected by the Illumina pipeline because of their poor match to the reference genome sequence. It would then be impossible for any subsequent alignment method to perform sequence variant detection with better sensitivity than that achieved by the pipeline.
File formats
Illuminator_data_extractor can extract data from the _prb.txt, fasta, _seq.txt and _qseq.txt file formats, and can generate _seq.txt formatted files. It also offers a facility for sorting sequence reads according to the sequence of a 5’ tag, writing sequences with identical tags into a tag-specific output file.
The program has two tabs: Sequence files and prb.txt files. The Sequence files tab converts fasta, _seq.txt and _qseq.txt formatted files to _seq.txt formatted files with the reads sorted according to their 5’ tag sequence. The prb.txt files tab handles the conversion of _prb formatted files to tag-sorted _seq.txt files. The mechanism for selecting which tags to use is the same for both tabs, and is explained below, with reference to the Tag selection panel on the first tab. If _prb files are to be converted, use the Tag selection panel on the prb.txt files tab.
Tag selection
If the sequence reads are to be sorted by 5’ tag, tick the appropriate check-box (highlighted by the green rectangle in Figure 1). This will enable the button, the 'Tag length' list box and the button (all above the green line in Figure 1). The maximum tag length is set at 6 nucleotides, which allows a theoretical number of 4096 unique tags. However, due to errors in the synthesis of the tag sequences and basecalling the sequence data, it may be advisable to have each tag differ by at least 2 positions; this will minimize the assignment of reads to biologically incorrect 'bins'.
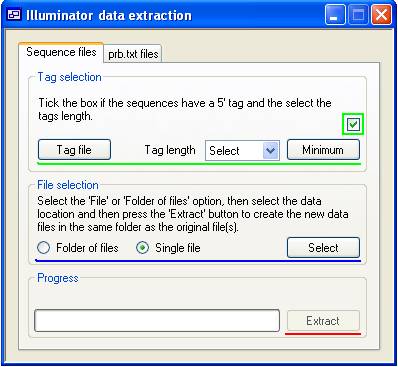
Figure 1
If you have a pre-existing list of the tags' sequences, you can load these using the button, which will allow you to select a text file listing the tags. This file should contain one tag sequence per line, where each tag must be the same length and no longer than 6 nucleotides. If you don’t have a list of tags, the program can screen the sequence read files for the presence of all tags of a certain length. To do this, first set the tag length using the 'Tag length' list box. Due to base-calling or synthesis errors, it is probable that all theoretically possible tag sequences will appear in a read data set. Therefore, in order to minimize the writing of large numbers of output files containing spurious tags, use the button to specify the minimum number of sequences containing a single tag, that must be present before that tag is considered genuine. Since, in order to do this, the program must first identify which tags are present in the reads, automatically identifying the tags will increase the time taken for data extraction by approximately 25%, compared to using a file of tag sequences. However, it has the advantage that it may identify erroneously tagged sequences originating from a systematic error in the tag synthesis. The sequences linked to each tag are written into an output file named after the tag sequence and (for the _prb derived files) the quality cut-off value used to filter the sequences. (See below.)
Converting fasta, _seq.txt and _qseq.txt files
These file formats are converted to tag-sorted _seq.txt files via the Sequence files tab (Figure 1). Define the 5’ tags as described above, and then select the data source. Since the read data may be contained either in a single sequence file or distributed across multiple text files in a single folder, select either the ‘Folder of files’ or ’Single file’ option in the File selection panel (above the blue line in Figure 1), press the button and pick either the data file or folder of data files. Finally, press (above the red line in Figure 1). The data output file(s) will be created within the same folder as the original data. The progress of the file conversion is shown in the Progress panel, whose legend displays the name of the file currently being processed. The progress meter increments as the reads are processed, with a full deflection representing one million reads, after which the progress bar is reset to zero.
Converting _PRB.txt files
The prb.txt files tab (Figure 2) allows the conversion of _prb formatted files to either _qseq.txt or fasta formats. This process is very similar to the conversion of fasta, _seq.txt and _qseq.txt described above. However the _prb files contain the base-calling quality scores (four for each nucleotide position). The definition of these quality scores is given here. The quality for each position ranges from -40 to +40 where -40 very strongly suggests that that base is not the correct nucleotide for that position while +40 very strongly suggests that that is the correct base. When converting the data in _prb.txt files to sequences, a quality score may be specified, below which the position is not called. The program will allow 2 uncalled bases per read. On encountering a third uncalled base, the sequence is no longer extended; if it is longer than 30 bases (excluding any tag) it is written to file; otherwise it is discarded. (However, if any low-quality position is encountered within the tag sequence, the read will be discarded.) To select the quality score cut-off, pick from the 'Quality' list box above the green line in Figure 2.
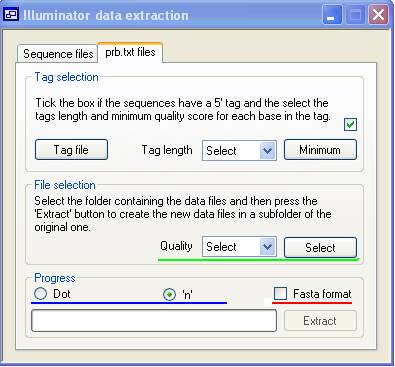
Figure 2
Since each lane of an Illumina run generates 100 _prb.txt files, the data is selected using the button (above the green line in Figure 2) to pick the folder containing the data files. Unlike extracting data from _qseq.txt, _seq.text and fasta files via the Sequence files tag it is possible to select if an uncalled base will be identified by the character ‘.’ or by ‘n’. Also, there is an option to convert the data to a fasta file ('Fasta format' check-box, above the red line in Figure 2). Pressing the button generates the sequence output data, with the progress indicated by the progress bar as described in the previous section.