Feature Guide
Table of Contents
2.1 Panels
2.2 Menu
2.3 Chromosome tabs
2.4 File list
3.1 Loading data
4.1 General layout
4.2 X-axis scale
4.4 SNP density
4.5.1 Effect of SNP density
4.5.2 Effect of isolated heterozygous SNPs and variable error rate
4.6 Flagging a specific map position
4.7 IBD cut-off
4.8 Physical position vs. genetic position
4.9 Comparison of patients with controls
4.9.1 Manually synchronizing data interfaces
5.1 Exporting composite IBD score images for all chromosomes
1. General description
In inbred individuals, mutations causing rare recessive disorders generally lie within an “autozygous” chromosomal segment that is identical by descent (IBD) from a common ancestor. IBDfinder is a program designed to rapidly identify IBD regions within patient Affymetrix SNP data. Since each patient’s SNP data file is analysed in isolation, patients need not be related to each other, originate from the same ethnic group, nor be genotyped using the same type of SNP chip. IBD regions are identified as stretches of chromosome devoid of heterozygous SNPs, and are scored according to the number of SNPs they contain. Where SNP density is very low, less reliance can be placed upon the IBD status of an apparently homozygous region. While this is not generally a problem for sets of 50k SNPs or more, it can be an issue with 10k SNP data. IBDfinder therefore demotes the IBD scores of regions that have less than a minimum SNP density of five per Mb/cM.
The general difficulty in using SNP data for autozygosity mapping is the limited heterozygosity (<0.5) of each individual marker. To infer the likelihood that a region is actually IBD, we use a simple concept; the number of homozygous SNPs that separate a given point from its nearest flanking heterozygous SNPs can be used as an index of the improbability of unnoticed heterozygosity. For example, if a SNP is flanked on both sides by 100 homozygous SNPs, the chance that it is in a heterozygous region is small compared to that for a SNP flanked by only 2 homozygous SNPs. Similarly, if a SNP is flanked by 5 homozygous SNPs on one side and 200 homozygous SNPs on the other, its chance of actually lying within a heterozygous segment is intermediate between the two previous examples.
To allow for the discontinuities that occur at the edges of IBD regions, two scores are generated for each SNP; the first is the smaller of the two distances (counted in homozygous SNPs) between it and its nearest flanking heterozygous SNPs. The second is the average of these two distances, a parameter that reflects the number of homozygous SNPs in the whole IBD region while keeping the same maximum value as the first parameter. These two scores will be referred to below as the and the scores, respectively.
2. Interface layout
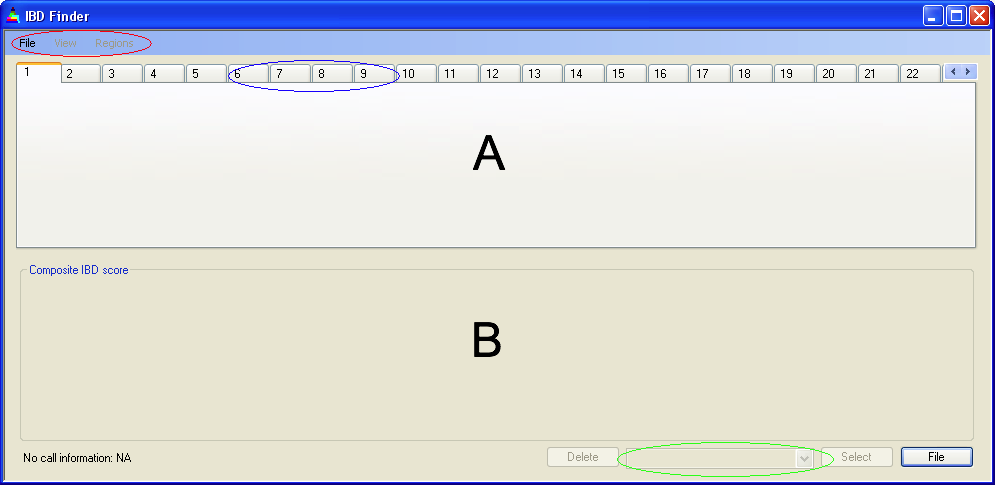
Figure 1: Program interface before data entry
The IBDfinder user interface consists of two main regions (A and B in Figure 1) and three main sets of controls (indicated by the red, blue and green ellipses).
2.1 Panels
A: This area is used to display the IBD scores of the selected data file and chromosome.
B: This region plots the number of files that show IBD at positions along the selected chromosome.
2.2 Menu
This contains three submenus: , and . (Figure 1, red ellipse.)
:
: This item allows you to open Affymetrix data files; it duplicates the function of the button in the lower right hand corner of the window.
: This item is enabled after the first file is successfully entered, and allows loading of all the files in a folder. Any file in the folder that is already loaded is ignored. A file will also be ignored if it does not contain the same type of positional map data as the first file loaded.
: This allows you to delete the current SNP genotype data. The button (bottom, centre right) duplicates this function.
: This closes the program.
and :
These menus control the manner in which the data are displayed, analysed and exported. Each function is described below.
2.3 Chromosome tabs
See Figure 1, blue ellipse. These are used to select the current chromosome, either with a mouse click or by using the ← and → arrow keys.
2.4 File list
This list (Figure 1, green ellipse) contains the names of the files currently loaded into IBDfinder. Selecting a file from this list (either using the mouse or via the ↑ and ↓ arrow keys) causes Panel A to display the IBD scores for that file.
3. Genotype data analysis
3.1 Loading data
Pressing the button at the bottom right or selecting the submenu allows the user to select an Affymetrix data file. These have a *.xls file suffix, but are in fact plain tab-delimited text files. [If IBDfinder cannot open your *.xls files, try opening them in Excel and resaving them as Text (Tab delimited) (*.txt), and then change the new file's extension to .xls.]
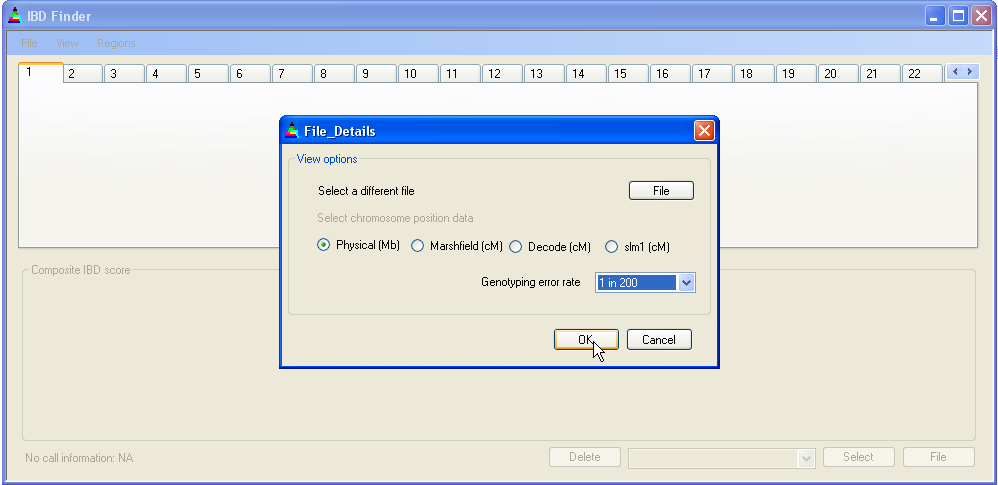
Figure 2: Entering the first file
Once a file is selected, IBDfinder queries it for positional data and allows you to select which type of map to use. Figure 2 shows a file containing all four positional data sets; however, if all of these are not represented in the file, only those that are present will be made available for selection. Since the positional data can be either in Mb or cM, distances will be referred to in this document as Mb/cM. The differences between physical and genetic distance are discussed below, in Section 4.8 Physical position vs. genetic position. The box also allows a different file to be selected (via the button) and the genotyping error rate (explained below) to be set via a drop-down list.
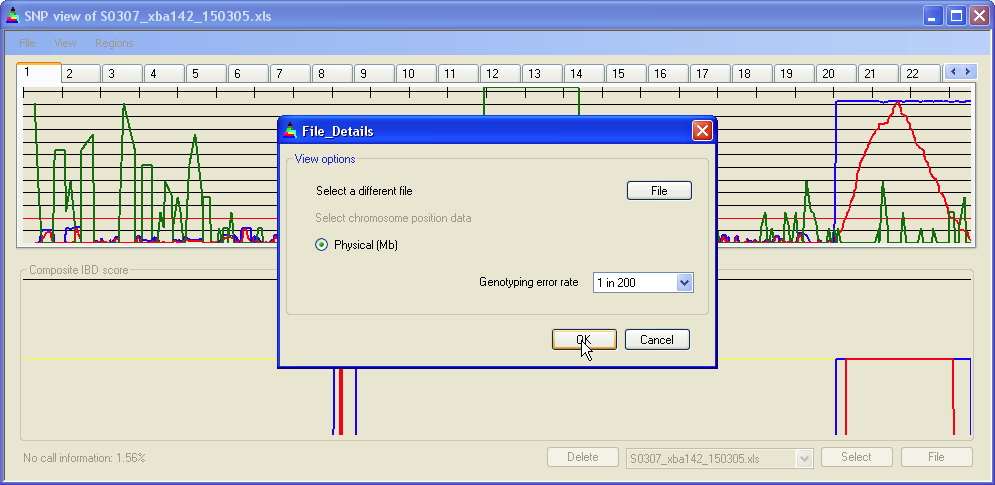
Figure 3: Entering further data files
Since an important design goal of IBDfinder was to allow comparison of data derived from different SNP sets (e.g. XbaI vs. HindIII), each input file has to include its own positional data. Also, the same type of positional unit (e.g. physical Mb) must be used for all files under comparison. To enforce this constraint, only the positional data type that was chosen for the first file is available for subsequent files; thus in Figure 3 only the radio button is available when loading new files. Once one file has been entered, the submenu is enabled, allowing the user to enter all files in a folder, using the same settings applied to the last file entered manually. [N.B. map positions in Affymetrix data annotation files change over time!! Check the uniformity of all positional data if multiple input files are being used!!]
3.2 Genotyping error rate
Affymetrix per-SNP genotyping accuracy is stated to be 99.4% to 99.6%, which implies that within an IBD region containing 200 SNPs, one SNP would be expected to be miscalled as heterozygous. To allow IBDfinder to accomadate this, the user can select an error rate (Figure 4), which is then used to determine whether to disregard heterozygous SNPs surrounded by extended runs of homozygous SNPs. This function is explained in more detail in the IBD score adjustment section.
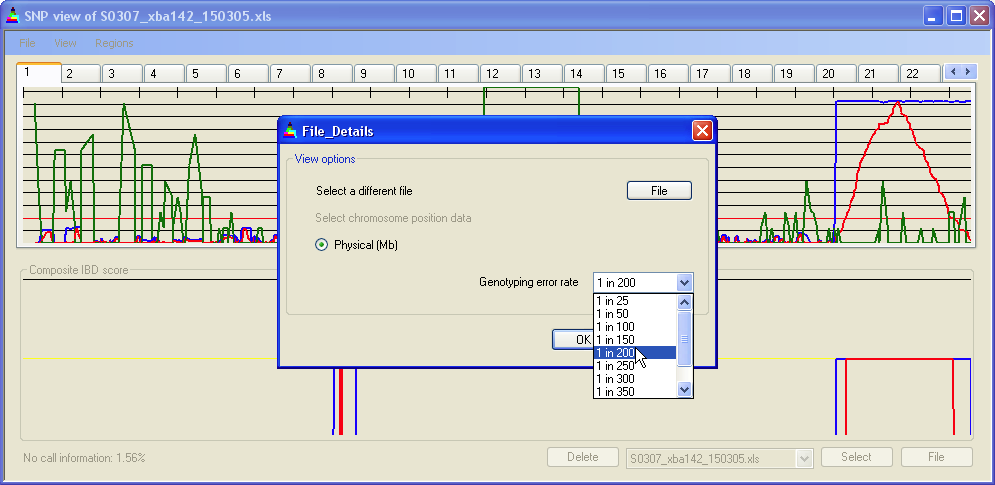
Figure 4: Selecting the genotyping error rate
4. Data display features
4.1 General layout
IBDfinder displays SNP data one chromosome at a time, with the currently selected file's IBD scores plotted in Panel A and the cumulative IBD scores in Panel B. The chromosomal position is represented along the X axis, each tick (Figure 5, D) marking 10 Mb/cM. The IBD score is plotted on the Y axis. The current file name is shown in the list box (Figure 5, red ellipse) and the current chromosome is identified by the yellow tab (Figure 5, blue ellipse). The percentage of “NoCall” SNPs for the current data file is also shown (green ellipse); this is a rough index of data quality. If the “NoCall” rate exceeds 5%, this value is displayed in red
In the upper panel, the IBD score is plotted as a blue line (Figure 5, A) and the score as a red line (Figure 5, B). The gridlines labelled F in Figure 5 indicate the magnitude of the score, at intervals selectable via the menu item.
The green graph (Figure 5, E) shows regions where the SNP density falls below 5 SNPs per Mb/cM. The way in which this is determined is outlined in the SNP density section.
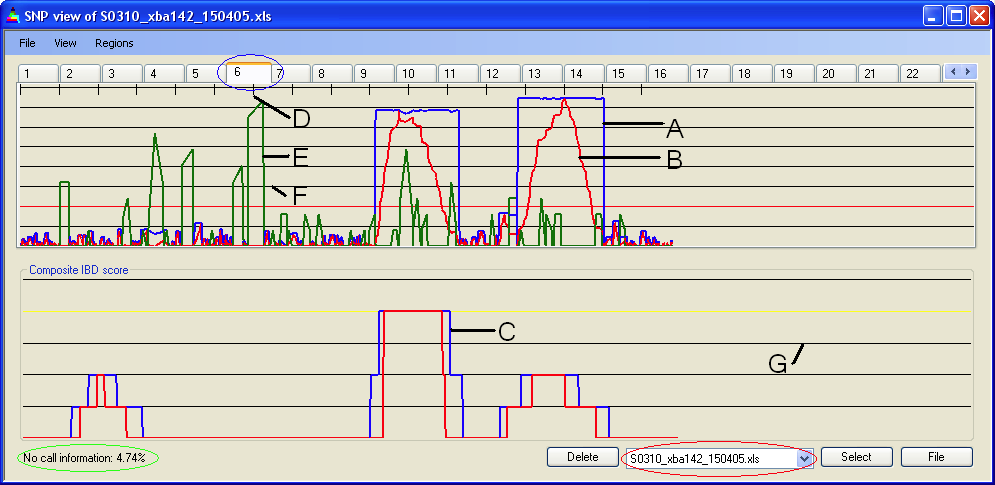
Figure 5: Data display
The cumulative IBD score is shown in the lower panel. This is calculated by dividing the chromosome into segments of 0.125 Mb/cM in length, which are then scored as IBD or not IBD, as follows:
Region is not IBD if:
- It contains a heterozygous SNP, irrespective of the number of homozygous SNPs in the segment.
- It does not contain any SNPs with an IBD score above the cut-off value.
- It does not contain any SNPS, and one of its flanking segments is not IBD.
Region is IBD if:
- It contains a SNP that has a score greater than the cut-off value.
- It does not contain any SNPS and both of its flanking segments are IBD.
The number of patients that are IBD for a segment is plotted on the graph in the Panel B. The Y axis shows the number of patients with IBD (labelled G in Figure 5); the yellow line defines the maximum score. Except where stated to the contrary, the pictures in this user guide illustrate the data from only one file.
4.2 X-axis scale
The X-axis scale can be adjusted so that the chosen chromosome is either stretched across the full length of the axis () or drawn to the same scale as Chromosome 1 (Figure 6a,b).
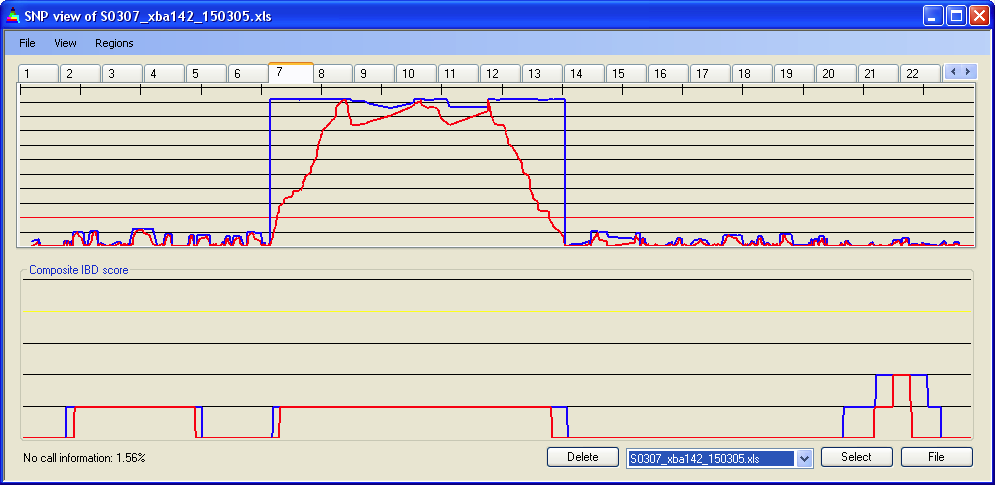
Figure 6a: SNP data for chromosome 7, scaled to the full length of the X axis
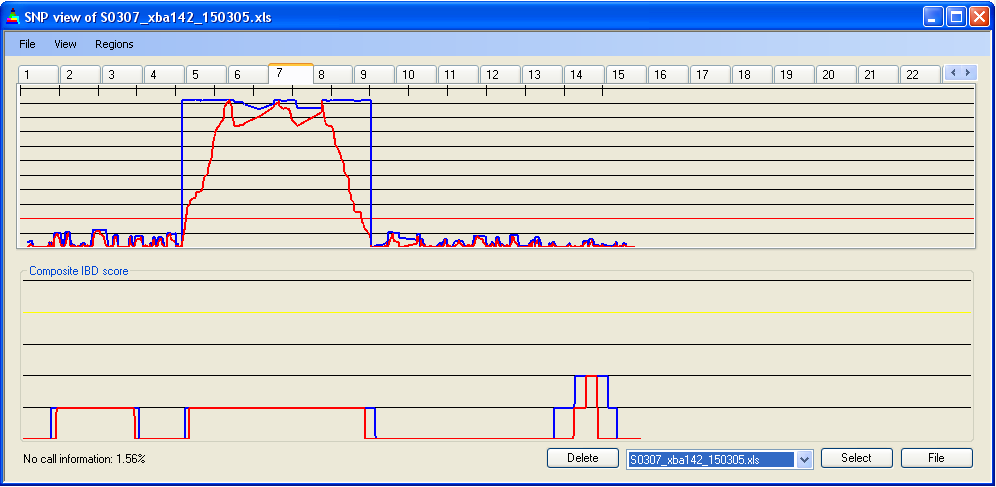
Figure 6b: SNP data for chromosome 7, to the same scale as Chromosome 1
4.3 SNP genotype data
Selecting from the menu displays the positions of homozygous SNPs as steel grey vertical lines (Figure 7a). Since the number of SNPs in a region can exceed the number of pixels representing it on the screen, multiple SNPs may be drawn on the same pixel. Therefore, this option should be used only to identify regions of low SNP density, not for counting SNPs. Selecting displays heterozygous SNPs as yellow vertical lines (Figure 7b). Both options can be selected simultaneously; however, since heterozygous SNPs are more useful when identifying IBD regions, the heterozygous SNPs (yellow) are drawn overlying any homozygous SNPs at the same screen position (Figure 7c).
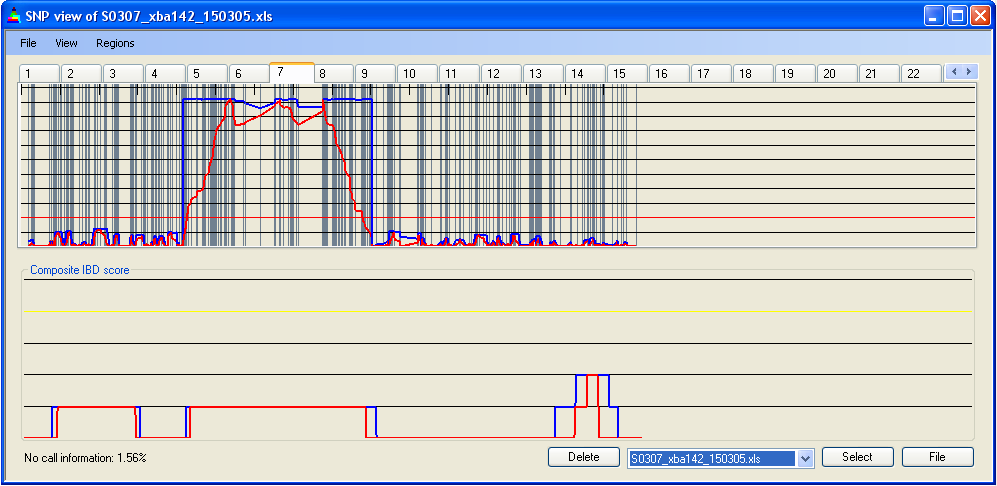
Figure 7a: Highlighting homozygous SNPs
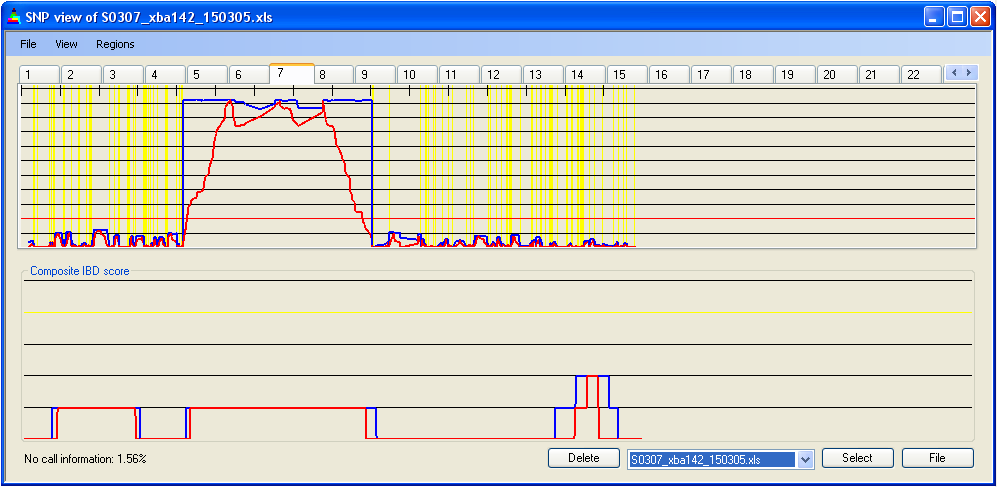
Figure 7b: Highlighting heterozygous SNPs
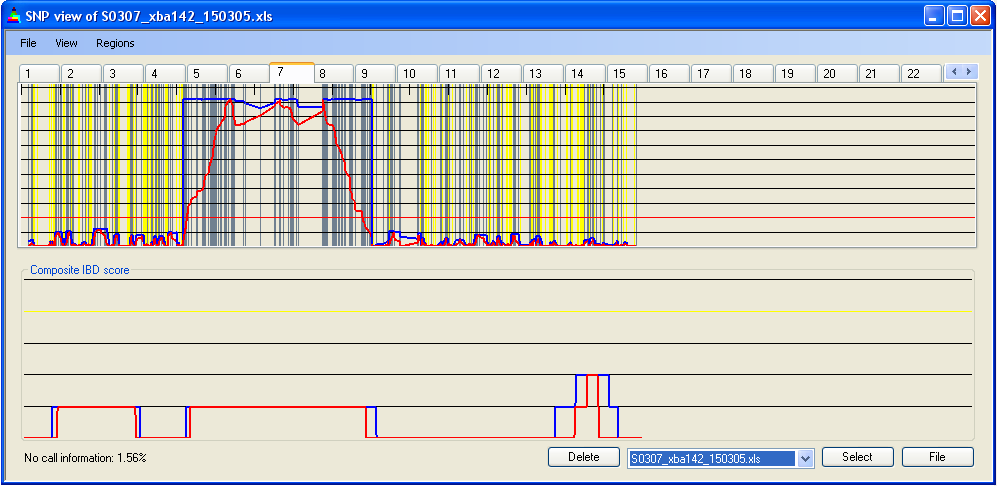
Figure 7c: Highlighting homozygous and heterozygous SNPs
4.4 SNP density
One of the assumptions made when IBDfinder analyses data is that there are at least 5 SNPs per Mb/cM. The option displays regions of low SNP density.
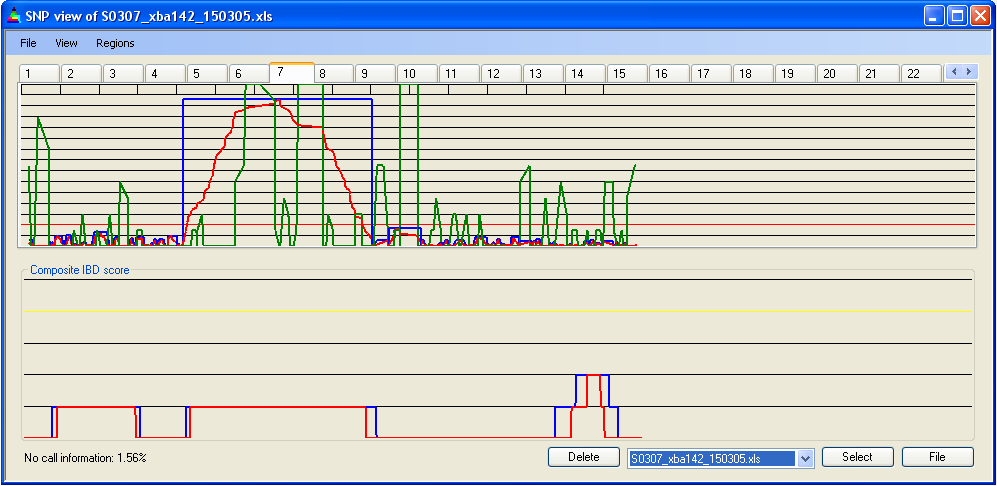
Figure 8: Identification of regions of low SNP density
For each SNP, the distance between its flanking SNPs is calculated. If this distance exceeds 0.6 Mb/cM, the region is deemed to be of low SNP density. In this case, the notional number of missing SNPs is determined; e.g. a region of 1 Mb/cM is expected to contain five SNPs, so if it only contains three, it is scored as missing two SNPs. If it is within an IBD region, then the number of missing SNPs is deducted from the IBD scores. The missing SNP score is plotted in green on Panel A, with a Y axis maximum value fixed at 10 (Figure 8). Generally speaking, this only significantly affects borderline IBD regions, or those that span the centromere (when using physical map distances). Otherwise, regions of low SNP density are a significant feature only when analysing data from 10k SNP chips. For a comparison of how genetic and physical distances change the apparent SNP density and location, see Physical position vs. genetic position.
4.5 IBD score adjustment
4.5.1 Effect of SNP density
As described above, local SNP density can be used to adjust the IBD scores. This feature is enabled by default (Figure 9a), but can be disabled by deselecting (Figure 9b). As seen by comparing Figures 9a and b, this adjustment can make a noticeable difference to the extent of IBD regions. This effect does not necessarily imply that a region is not IBD, but does indicate that the data on which the decision is based are less strong than for other regions.
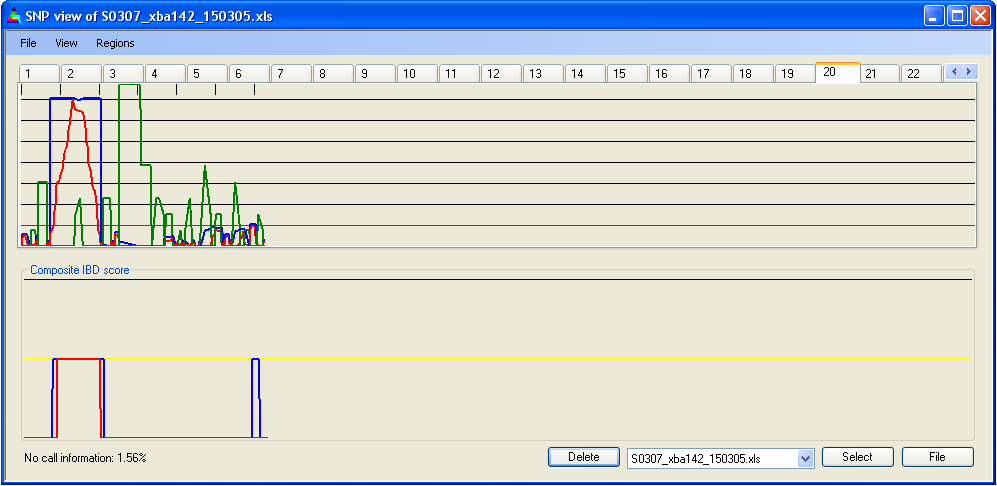
Figure 9a: Adjusted IBD scores
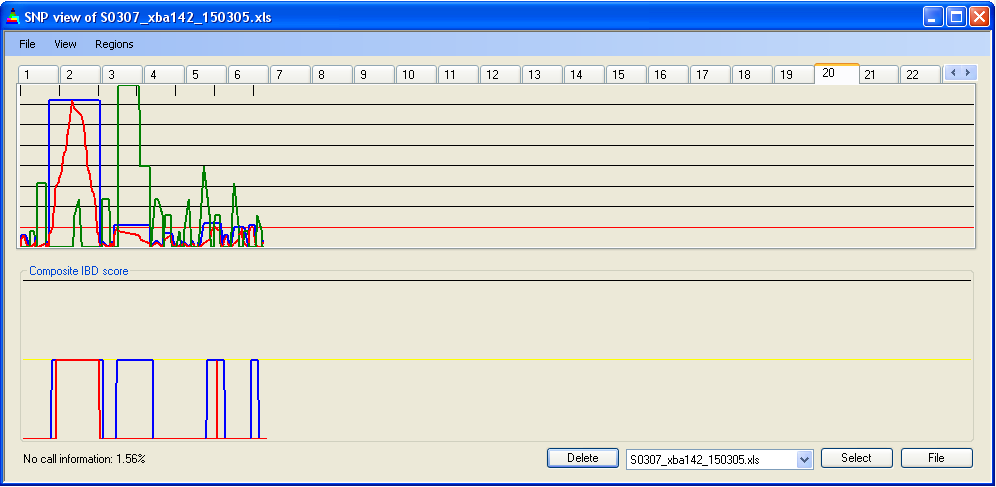
Figure 9b: Unadjusted IBD scores
4.5.2 Effect of isolated heterozygous SNPs and variable error rate
As mentioned above under data entry, there is an approximately 1 in 200 chance that a homozygous SNP is miscalled as heterozygous. At the time of data entry, the user can select an error rate between 1 in 25 and 1 in 450. This can then be used to test the significance of a single apparently heterozygous SNP within a long stretch of homozygous SNPs. If a 1 in 100 error rate is selected, a heterozygous SNP is regarded as an erroneous call if it lies within a homozygous region containing >99 SNPs and is more than 100/3 SNPs from the nearest heterozygous SNP. (This means that a region of more than 100 SNPs could have multiple miscalled SNPs, but only if they are 34 or more SNPs apart.) To highlight the uncertainty of the IBD score in the area surrounding the suspicious SNP, the IBD score is reduced to the IBD cut-off value (see IBD cut-off below).
Figure 10 shows SNP data from a 50k SNP chip, highlighting four regions that reach the IBD cut-off value. The first two of these regions are bordered by two separate points (1 and 2 in Figure 10a) at which heterozygous SNPs occur; the last two are separated by one such point (3 in Figure 10a). However, when the IBD scores are adjusted (Figure 10b) these regions merge to form two large zones of IBD. When the data are entered under a more stringent error rate of 1 in 450, this does not occur (Figure 10c).
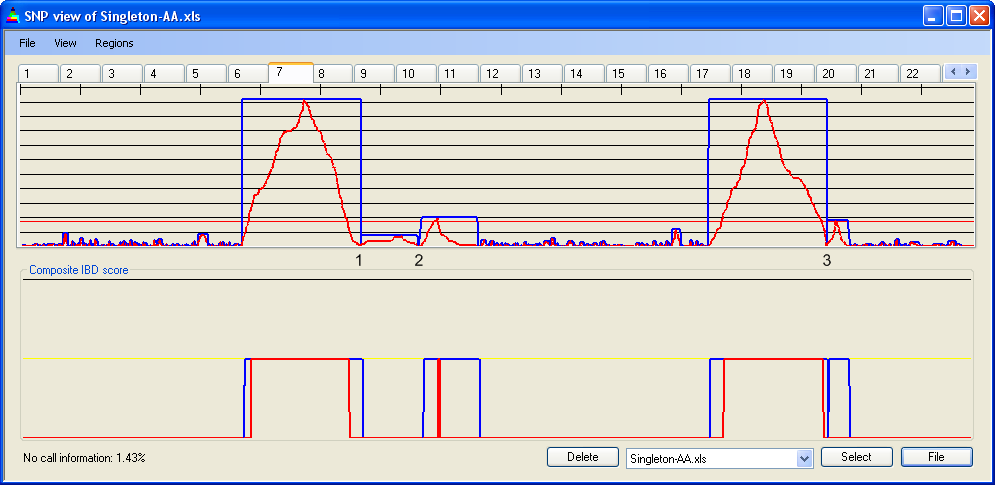
Figure 10a: Unadjusted IBD scores for chromosome 7: 50k SNPs, error rate 1/100
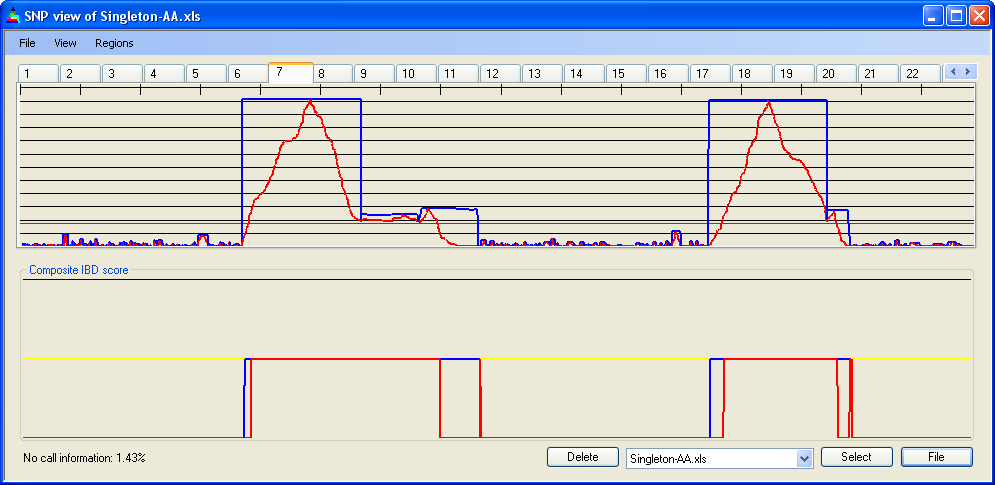
Figure 10b: Adjusted IBD scores for chromosome 7: 50k SNPs, error rate 1/100
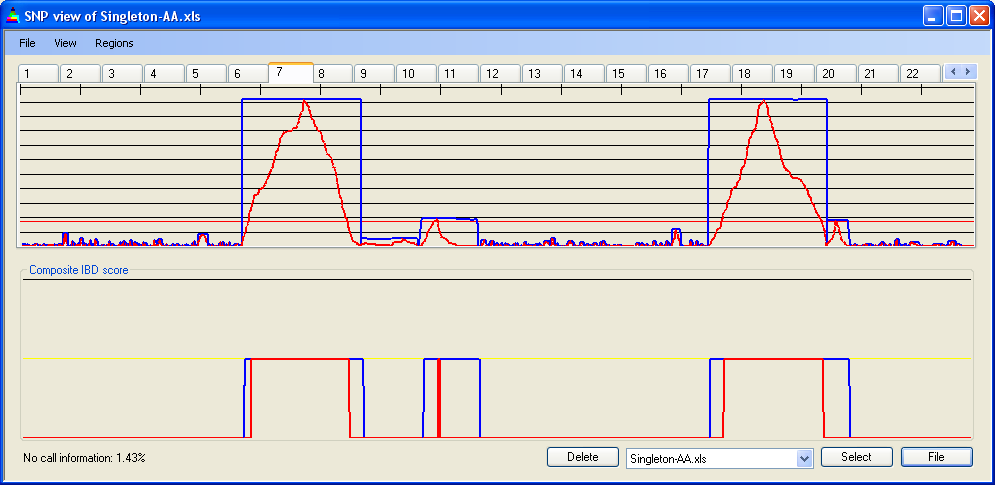
Figure 10c: Adjusted IBD scores for chromosome 7: 50k SNPs, error rate 1/450
4.6 Flagging a specific map position
The location of a candidate gene can be overlaid onto the IBD data, as a black vertical line, by selecting the menu item (Figure 11) and entering that gene's position, in the same units as those used to display the SNP data.
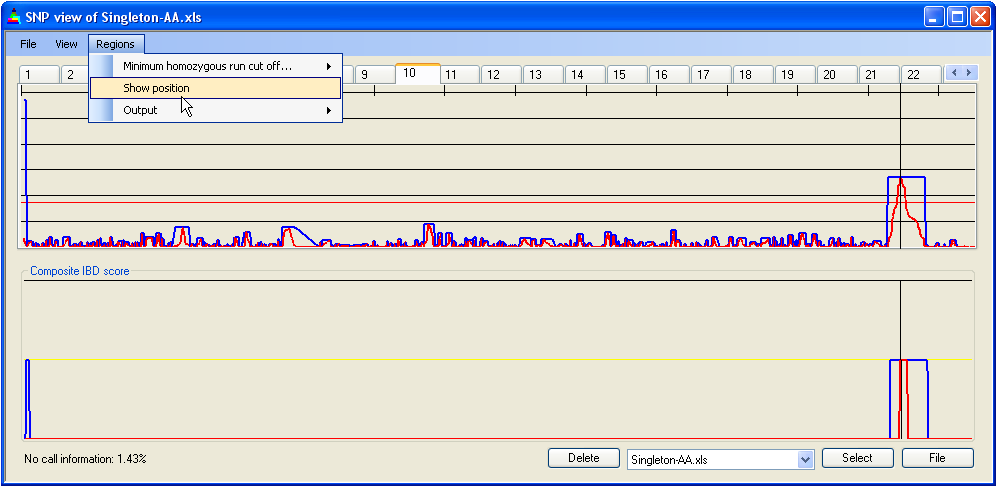
Figure 11: Flagging a specific point along the chromosome
4.7 IBD cut-off
The minimum run of homozygous SNPs needed to define an IBD region can be set via . The current value is displayed as a red line across the Panel A graph. In Figures 12b-d, the cut-off has been set to 5, 15 and 30 homozygous SNPs respectively. As the cut-off value changes, the cumulative IBD score for the four patient genotype files (in Panel B) can be seen to reduce. The spikes of IBD in Figure 12b are likely to result both from chance occurrences of runs of >5 homozygous SNPs and SNPs in linkage disequilibrium.
The homozygous run cut-off should be determined empirically, since the number of consecutive homozygous SNPs that can occur by chance depends on the level of background inbreeding in the patients’ population. Analysis of SNP data from the outbred CEPH families suggests that between 68% and 71% of SNPs are homozygous; however for consanguineous individuals this will be elevated. Table 1 shows the approximate number of runs of homozygous SNPs expected in a 50k SNP data set for different levels of homozygosity, assuming no linkage disequilibrium.
| Consecutive homozygous SNPs |
Number of occasions that a run of homozygous SNPs occurs by chance at different levels of homozygosity | |||
|---|---|---|---|---|
| 65% | 70% | 75% | 80% | |
| 1 | 32500 | 35000 | 37500 | 40000 |
| 2 | 10563 | 12250 | 14063 | 16000 |
| 3 | 4577 | 5717 | 7031 | 8533 |
| 4 | 2231 | 3001 | 3955 | 5120 |
| 5 | 1160 | 1681 | 2373 | 3277 |
| 6 | 628 | 980 | 1483 | 2185 |
| 7 | 350 | 588 | 953 | 1498 |
| 8 | 199 | 360 | 626 | 1049 |
| 9 | 115 | 224 | 417 | 746 |
| 10 | 67 | 141 | 282 | 537 |
| 11 | 40 | 90 | 192 | 390 |
| 12 | 24 | 58 | 132 | 286 |
| 13 | 14 | 37 | 91 | 211 |
| 14 | 9 | 24 | 64 | 157 |
| 15 | 5 | 16 | 45 | 117 |
| 16 | 3 | 10 | 31 | 88 |
| 17 | 2 | 7 | 22 | 66 |
| 18 | 1 | 5 | 16 | 50 |
| 19 | 1 | 3 | 11 | 38 |
| 20 | 0 | 2 | 8 | 29 |
Table 1: Occurrence of homozygous runs for different levels of homozygosity
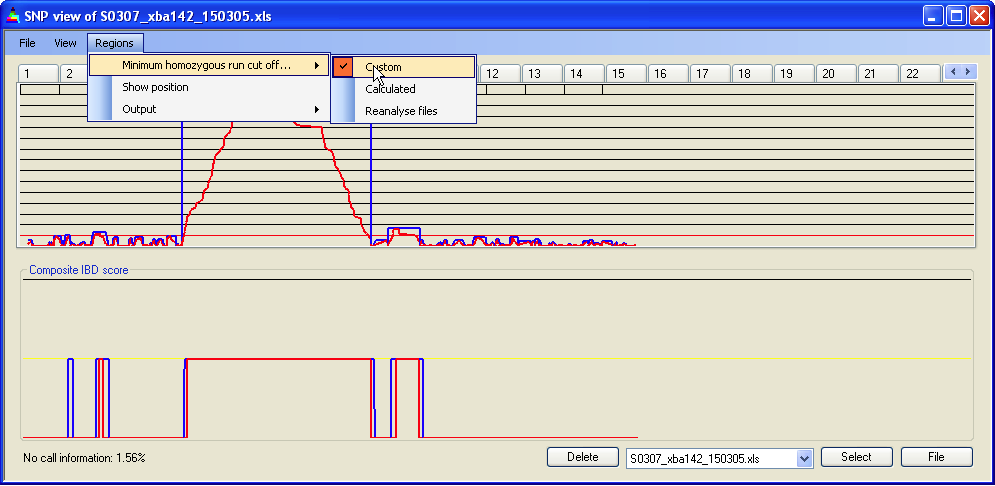
Figure 12a: Setting the minimum homozygous run cut-off value
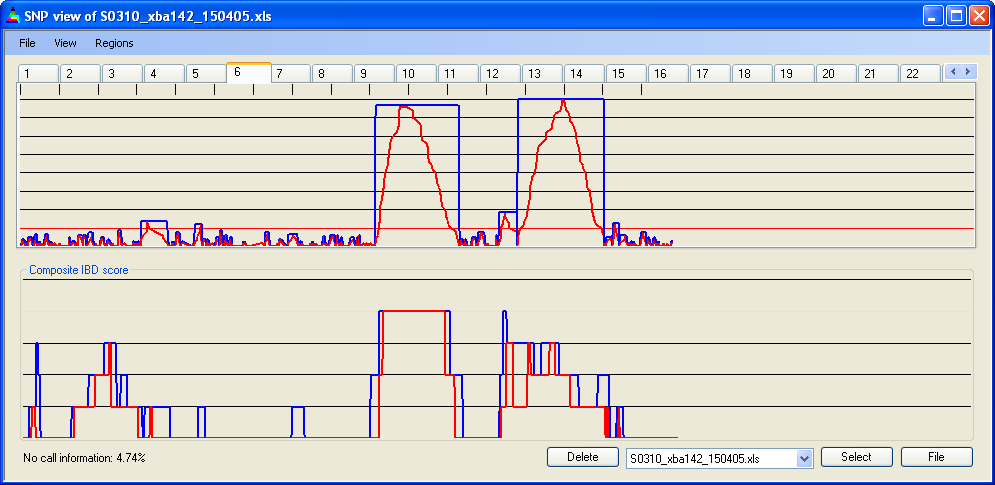
Figure 12b: Accumulated IBD score; four 10k SNP data files, cut-off value = 5 SNPs
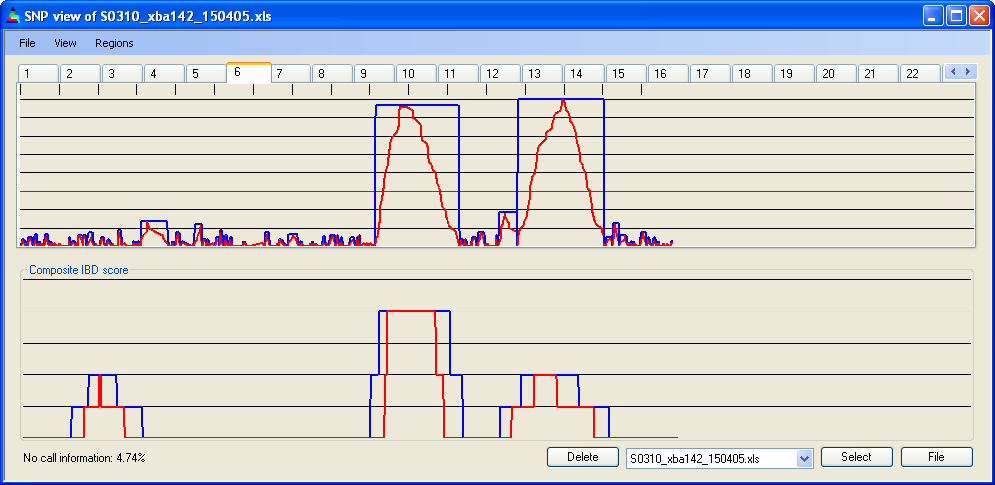
Figure 12c: Accumulated IBD score; four 10k SNP data files, cut-off value = 15 SNPs
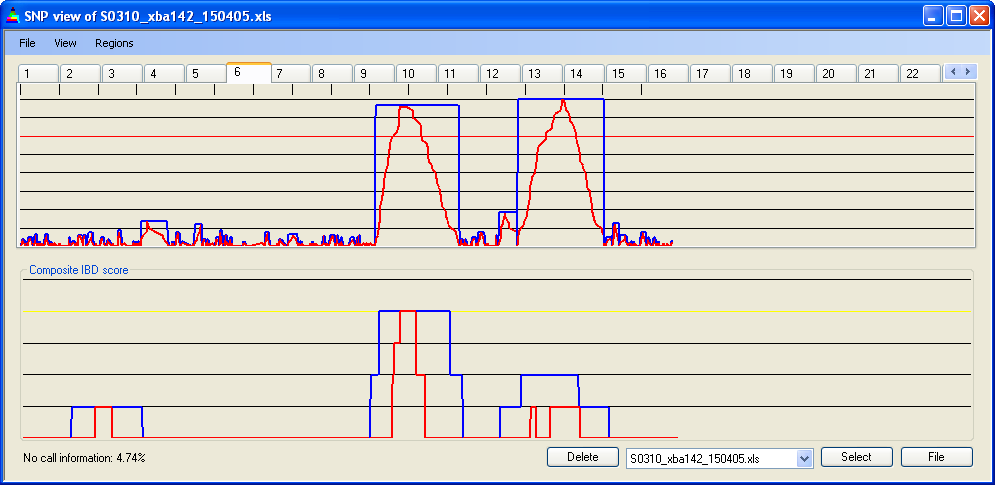
Figure 12d: Accumulated IBD score; four 10k SNP data files, cut-off value = 30 SNPs
4.8 Physical position vs. genetic position
SNP data can be aligned by either genetic or physical distance; since the rate of recombination varies along a chromosome the two methods can produce different results, especially if the data are adjusted for SNP density. Figures 13a-d show SNP data aligned by either physical or genetic position and clearly shows the different distribution pattern. Due to the lower recombination rates around centromeres and heterochromatic DNA, the pericentromeric gap in the SNP distribution on Chromosome 1 (Figure 13a) almost completely disappears when the data are aligned by genetic position (Figure 13b).
Similarly, the increased recombination rate towards the telomeres and the presence of recombinational hotspots may cause SNP densities to differ when measured in physical (Figure 13c) or genetic (Figure 13d) units. This difference can have a marked effect when IBD scores are adjusted by SNP density; see for example the identification of a small region of IBD when viewed by physical but not genetic distance (Figure 13c,d). However, this is not normally a problem with 50k or larger SNP data sets.
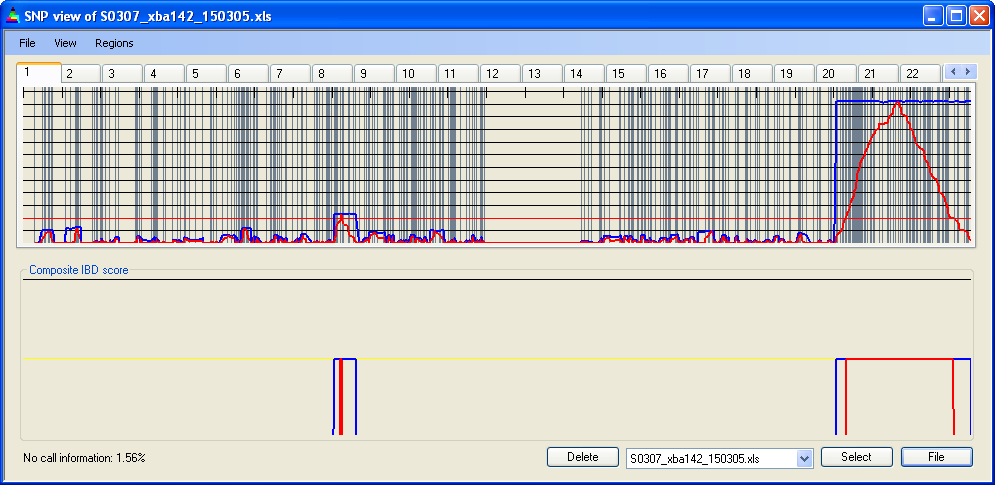
Figure 13a: IBD scores and location of homozygous SNPs aligned by physical position
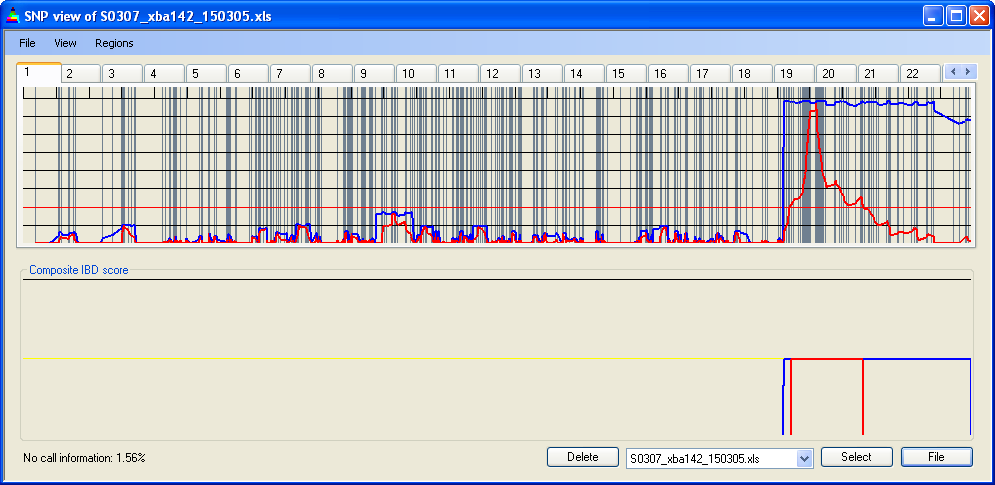
Figure 13b: IBD scores and location of homozygous SNPs aligned by genetic position
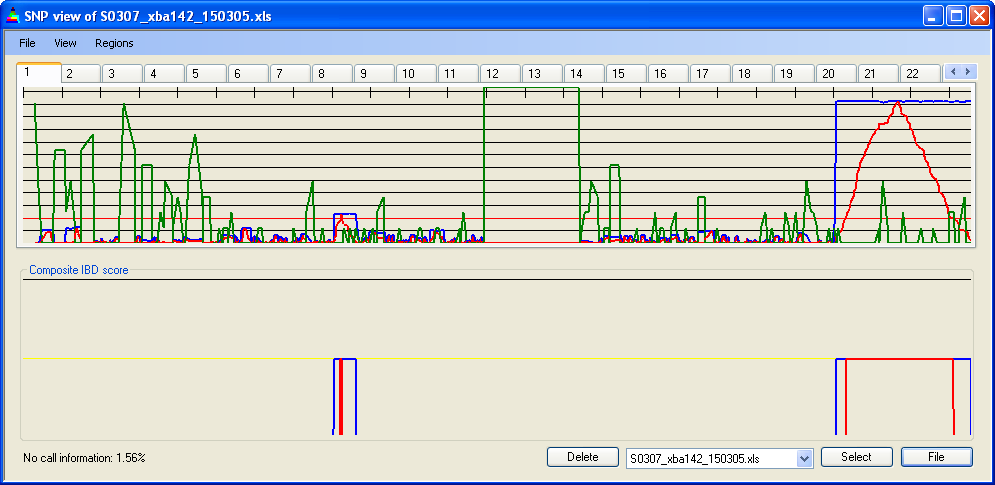
Figure 13c: IBD scores and SNP density aligned by physical position
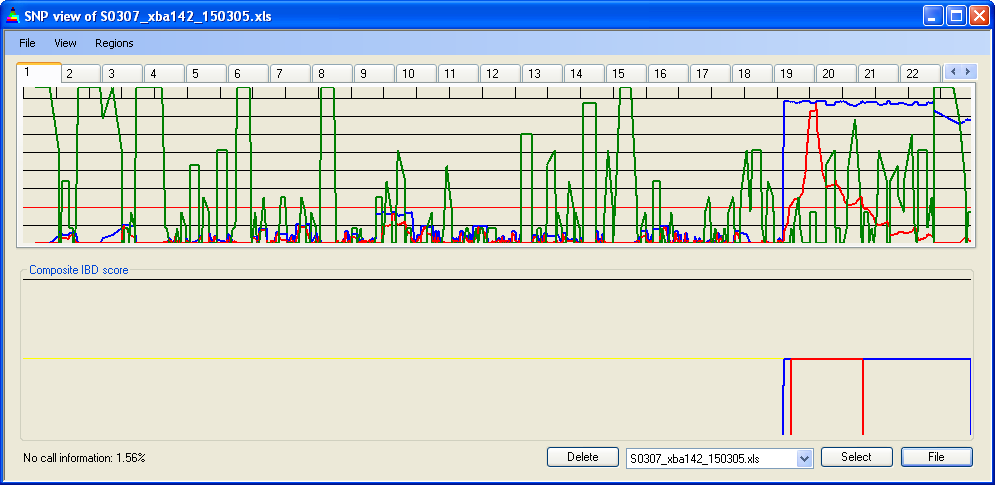
Figure 13d: IBD scores and SNP density aligned by genetic position
4.9 Comparison of patients with controls
Once patient data have been loaded, they can also be compared to data from control or unaffected individuals. To do this, click the menu item (highlighted in Figure 14 by the turquoise ellipse). This opens a second window that contains a panel equivalent to the lower panel in the main window. Control data are added to the form via the menu, in the same way that data are added to the main form. By default, changes applied to the patient data are automatically applied to the control data; however this behaviour can be suspended, as shown below. Closing the control data window will discard all data loaded into it.
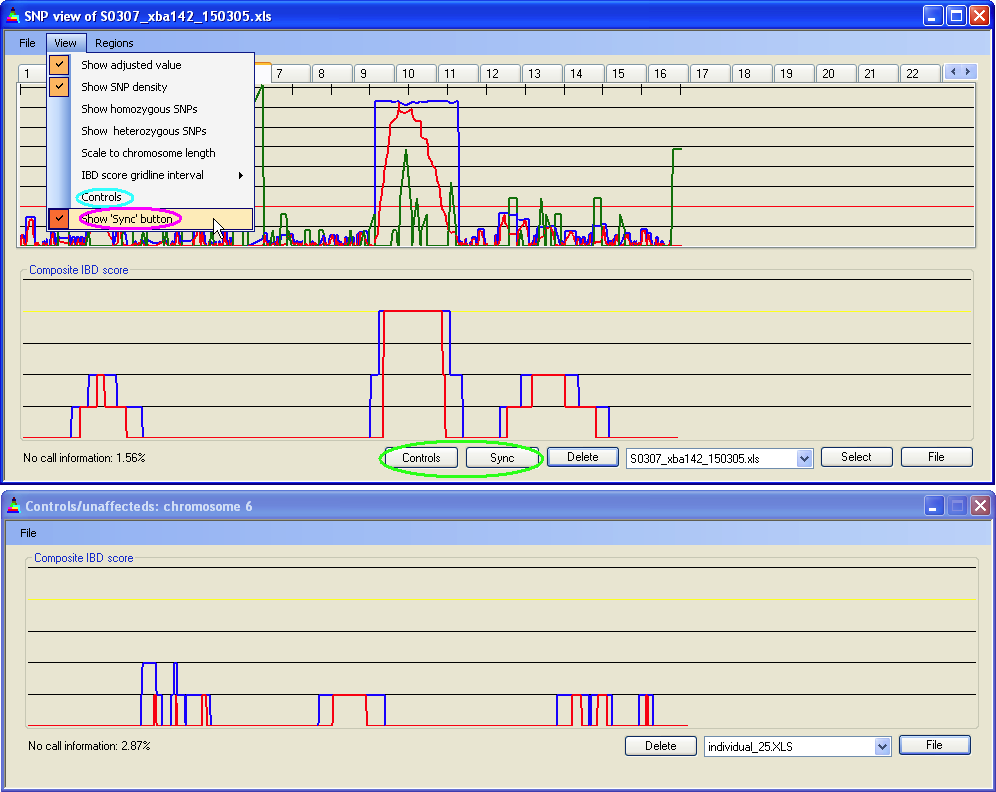
Figure 14: Data in IBDFinder can be compared to control data to identify common regions of homozygosity found in unaffected individuals
4.9.1 Manually synchronizing patient and control data interfaces
When working with large data sets, it may become inconvenient to refresh the program each time the view is edited by either the or submenus. If the menu item is ticked (violet ellipse in Figure 14) the button will appear on the main window (green ellipse in Figure 14). If the control data form is present, a second button will also appear. Changes made via the menu will now not be implemented unless is pressed. However, changes made via the menu, or by changing the selected chromosome, will be applied to the main form, but not to the control data form. To update the control data, the user must click .
5. Exporting SNP data
The SNP data underlying an IBD region can be saved to a tab-delimited text file, by simply left-clicking with the mouse pointer over the region of interest. The type of IBD score to be saved is selectable via the submenu (Figure 15). (If the option is chosen, the exported SNP data set will be the same as for the export option, but both IBD scores are exported.)
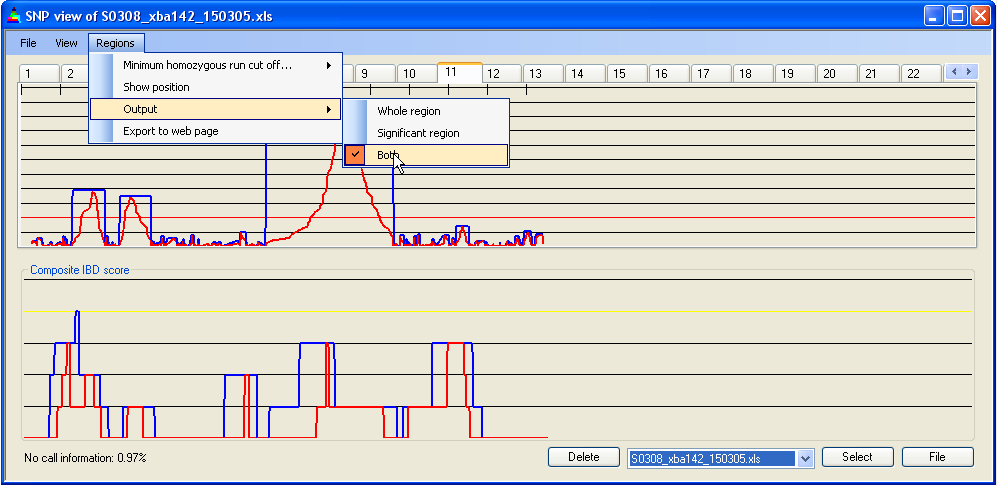
Figure 15: Selecting the type of IBD score data to export
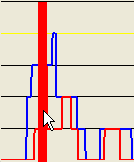
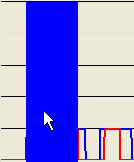
By clicking at different points within the lower panel, different subregions can be selected for data export, as shown below. Thus, with the pointer positioned as in Figure 16a, the red area in Figure 16b ( option) or the blue region in Figure 16c ( or options) will be exported, respectively.
 |
 |
 |
| a | b | c |
|---|
Figure 16: The extent of the SNP data region exported is mouse-selectable
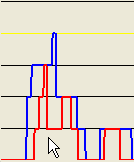
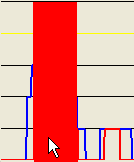
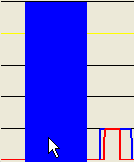
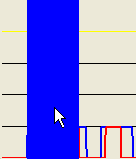
Alternatively, if a point closer to the baseline of the plot is selected (Figure 17a), the exported regions will be correspondingly broader, as shown in Figures 17b-c.
 |
 |
 |
| a | b | c |
|---|
Figure 17: A larger mouse-selected region for SNP data export
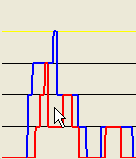
Figures 17b and c again correspond to the use of or options, respectively. If the pointer is clicked at a location outside the red scoreline (Figure 18a), data will be exported only if the option is selected (Figure 18b). (No data would be exported with the option selected.)
 |
 |
| a | b |
|---|
Figure 18: The extent of the SNP region exported by clicking outside the red graph
Finally, if the pointer lies outside both the red and blue boundaries (Figure 19), no data will be exported, since the selected point does not correspond to a region with a score above the cut-off value for either or IBD scores.
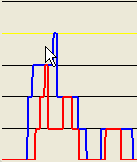
Figure 19: No data will be exported
5.1 Exporting composite IBD score images for all chromosomes
The standard IBDfinder window only allows data from one chromosome at a time to be viewed. To view the composite IBD scores for all chromosomes together, the menu item can be used. This will generate an HTML file containing a listing of all the composite IBD scores, in chromosome order. An example of such a page is here.
Note on positional coordinates in Affymetrix data annotation files
The integration of SNP data sets derived at different times, or from different SNP sets (e.g. 10k vs. 50k, XbaI vs. HindIII) is problematic, especially since positional data associated with individual SNPs may change with time. To deal with this problem, we have developed a sister application, SNPsetter, that will analyze a number of SNP data files for their marker content, and match or standardize them all to a common reference file containing marker locations. This is an important step in allowing data from different sources to be compared. The use of SNPsetter is described in a separate document.