Walk through guide
n.b. Earlier versions of this guide in PDF format are included in the AutoSNPa download bundles and refer to features of earlier program versions, some of which have changed.
1—Preliminary: data file considerations
An Affymetrix SNP data file is formatted as tab-delimited text. On Windows, such a file’s .xls file extension will cause it to be opened by Excel by default. However, AutoSNPa can only use the original flat text file, and cannot use data saved as an Excel workbook. If you have saved your data as an Excel file, first convert it back to tab-delimited text, via the menu option in Excel.
When viewed in a spreadsheet program, the data will be arranged in a number of columns, containing the allele frequencies, SNP position data, names and genotypes. The AutoSNPa program does not use most of this data, much of which is therefore superfluous. The minimum dataset required by the program is shown in Table 1. Each of these columns must contain the information indicated, while other columns can be deleted from the data file.
| Column | Data |
|---|---|
| 1st | SNP index |
| 2nd | SNP ID |
| 3rd | SNP database RS ID |
| 4th | Chromosome |
| 5th | Physical distance (bp) |
| 7th | Genotype data |
Table 1: Minimum file structure needed by AutoSNPa.
By default, SNP data are displayed with respect to physical position (5th column). If you wish to display markers relative to their Marshfield genetic positions, then the column entitled marshfield avg must also be included within the file, with its correct heading.
Every 3 months, Affymetrix updates its SNP information dataset, and removes SNPs that have failed quality control procedures. This may result in your Affymetrix files containing different SNP datasets, even if your samples were sent for analysis together. To ensure that all your data files use a common SNP data set it is advised that you reprocess the data with either the Affymetrix software or, if you do not have the cel file, with SNPsetter.
2—Creating the SNP database
2.1—Entering disease information
Before creating the AutoSNPa database, you may enter a disease ID number; name and phenotype information. This information is accepted using the button (Figure 1), and is used solely to verify that the correct file has been opened during subsequent data analysis.
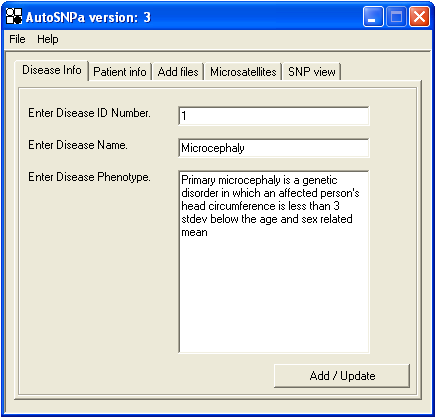
Figure 1: The AutoSNPa opening screen.
2.2—Addition of pedigree and patient information
When entering genotype data into AutoSNPa, it may be advantageous to link the data to individuals within a pedigree. Note, however, that this is no longer obligatory, and data files can be entered directly by omitting this step.
Pedigree and patient information can be entered either graphically or via the keyboard; both methods are accessed through the Patient info tab (Figure 2).
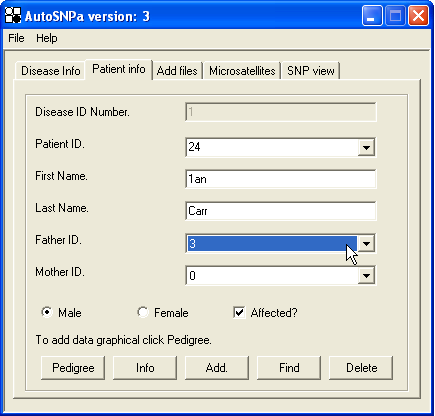
Figure 2: Patient info tab, used to enter an individual person into the database.
2.2.1—Pedigree construction using the keyboard
To construct the pedigree database using the keyboard, first enter the individual’s unique ID (which must be a digit), first name and last name, and then select their parents using the Father ID and Mother ID drop-down lists. (If either of the two parents does not yet exist in the database, enter “0” for that parent.) The Father ID and Mother ID lists only contain the ID numbers of individuals previously entered into the pedigree; to construct a pedigree in this way therefore requires parents to be entered before offspring. Although this constraint helps to reduce errors in pedigree construction, it can become difficult to construct complex consanguineous pedigrees in this manner.
An already entered individual’s information can be modified by selecting his or her ID number from the Patient ID drop-down list and clicking . If edited, the d will be updated after clicking (which was previously labelled ). Also, when a patient ID is selected from the list, the button summons a dialogue box giving a brief summary of the individual’s nuclear family and SNP genotype data. Individuals can be removed from the database using the button. However, this action does not remove that individual’s descendants, and so may result in incorrectly formed pedigrees that will not be drawn correctly.
2.2.2—Graphical construction of a pedigree
Clicking on the button of the Patient info tab opens a second form (Figure 3), which allows graphical pedigree creation. If the AutoSNPa database already contains patient information, this form will display the pedigree.
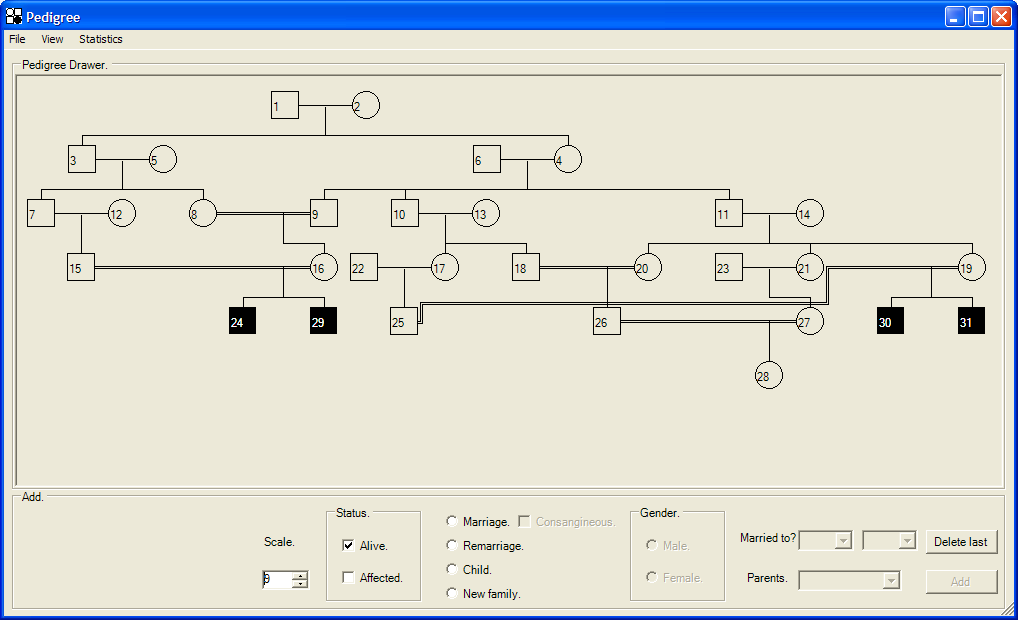
Figure 3: The Pedigree form, displaying a consanguineous pedigree.
2.2.2.1—Adding a new family
If no individuals are present in the database, the form shown in Figure 3 will be blank, in which case a founding couple must be added by clicking the New family radio button (Figure 4). It is possible to add more than one new family in this way, and the descendants of each family can marry.
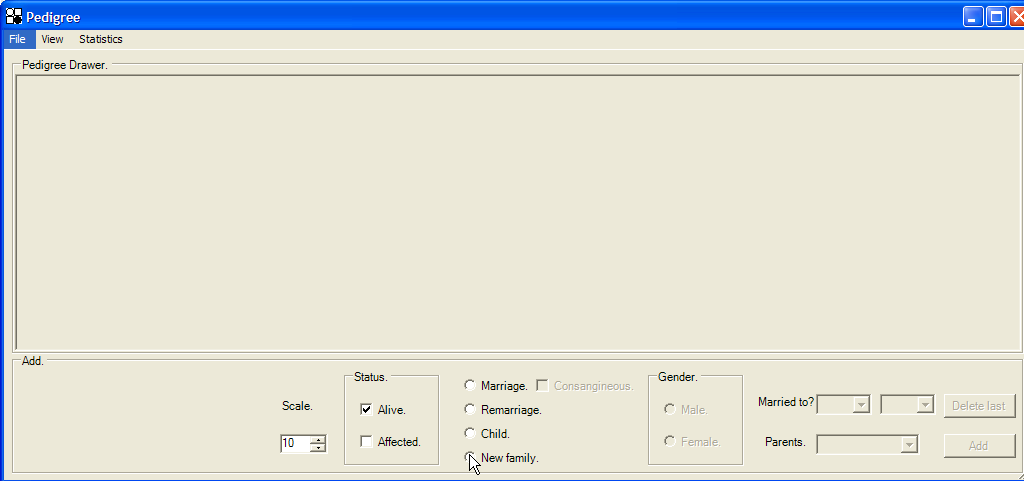
Figure 4: Addition of a new family.
2.2.2.2—Adding children
Offspring can be added once a founding couple has been defined. First, click on the Child radio button above the New family radio button. This enables the Gender radio buttons; after selecting the child’s sex, choose its parents from the Parents drop-down list. This list offers all marriages previously entered into the pedigree, each as two ID numbers linked by a hyphen. The first number is the husband’s ID and the second is the mother’s ID. Clicking the button (Figure 5) draws the new child. When adding a new individual, you can specify whether that individual is affected and/or alive. (This may also be done after the individual has been added, but can be a useful option now, especially when drawing the early part of a pedigree in which the majority of the individuals have died, or when adding an affected child to a large family.)
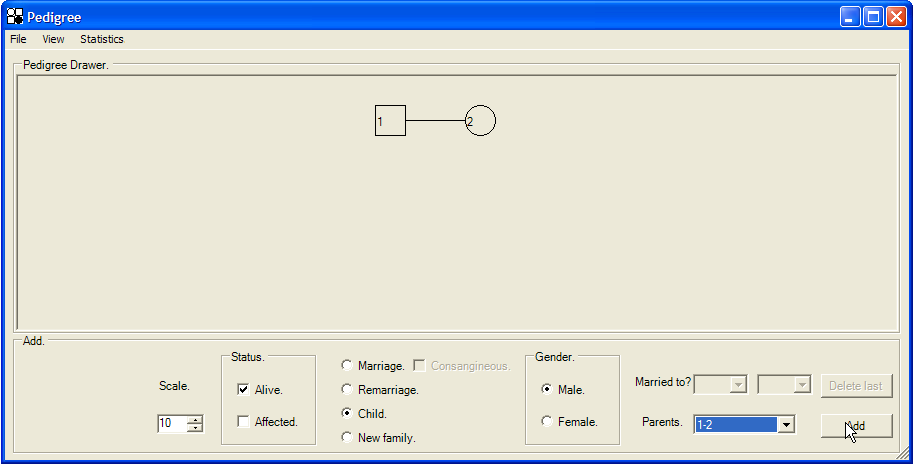
Figure 5: Adding a child. In this instance, a living unaffected male child will be added to the family of individuals 1 (father) and 2 (mother).
2.2.2.3—Adding marriages to the pedigree
Once children have been added, it is possible to add marriages to the pedigree. Click the radio button and select the sex of the new individual; this enables one of the two Married to? list boxes; e.g. when adding a female to the pedigree in this way, the left-hand box is enabled, allowing selection of any unmarried male already in the pedigree (Figure 6). The marriage is created by clicking the button. Children can be added to such new marriages in the same way as to the original founder family.
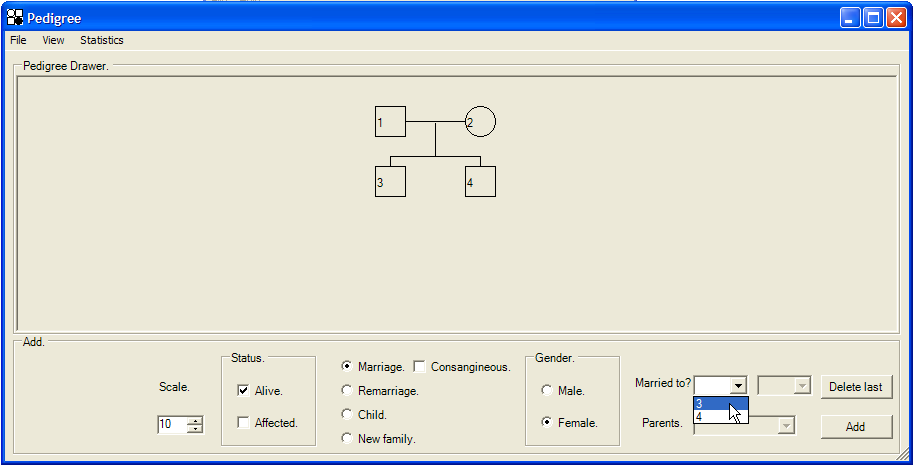
Figure 6: Adding a marriage to the pedigree. The figure shows the creation of a marriage between a new female and individual 3.
2.2.2.4—Adding a consanguineous marriage
As more individuals are added to the pedigree, it becomes possible to create consanguineous marriages. To do this, select the Marriage radio button and then check the Consanguineous check box next to it. This enables both Married to? list boxes. Use these to select the husband and wife and click to create the marriage (Figure 7).
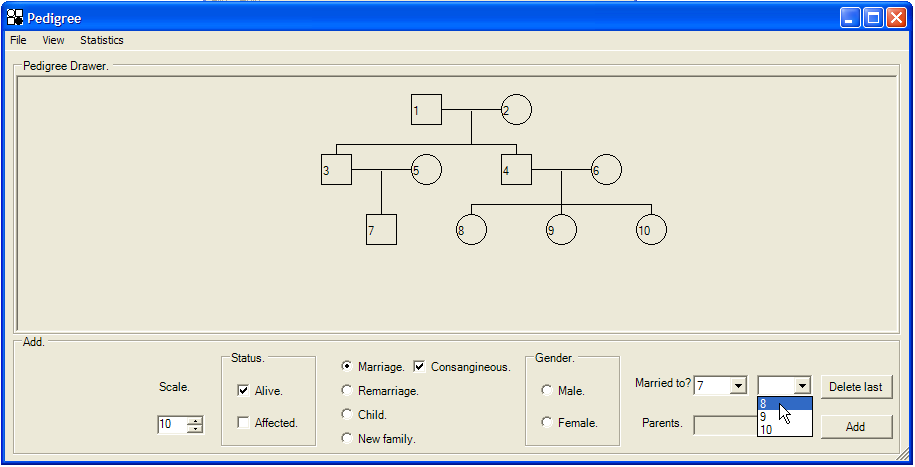
Figure 7: Adding a consanguineous marriage. The figure depicts the addition of a consanguineous marriage between husband 7 and wife 8.
When children are added to a consanguineous marriage, they always appear to the left of the right-most parent (Figure 8).
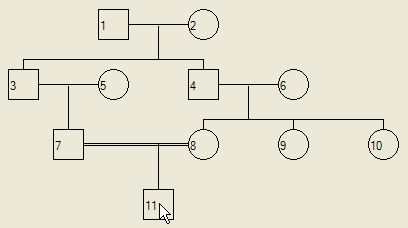
Figure 8: Children of consanguineous marriages are drawn to the left of the right-hand parent.
2.2.2.5—Adding second marriages
To add a second marriage to the pedigree, click the Remarriage radio button, to display the form shown in Figure 9. Select the person you wish to remarry and then click the New Person radio button, followed by . This will add a new individual to the pedigree, married to an individual who is already married. (If you select an individual who is not already married, then that person will become married, just as if you had selected the Marriage radio button described earlier.)
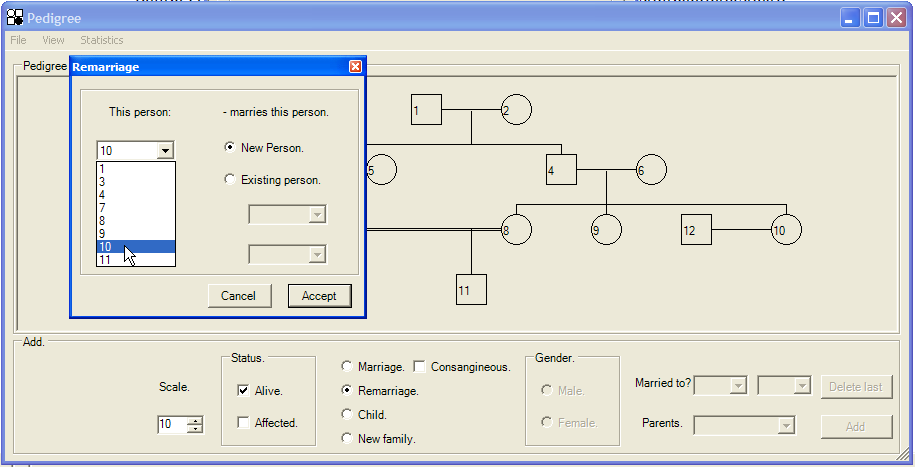
Figure 9: Adding a second marriage to the pedigree. The figure shows the addition of a second marriage between individual 10 and a new individual.
It is also possible to create a second marriage between individuals already present in the pedigree. This is done as described above, but by selecting the Existing person radio button rather than New Person, and then choosing a partner from the enabled list box (Figure 10). The only limitation to forming remarriages is that at least one of the individuals must be genetically related to other members of the pedigree (i.e. in Figure 11, individual 6 could not remarry individual 13). WHY IS THIS??
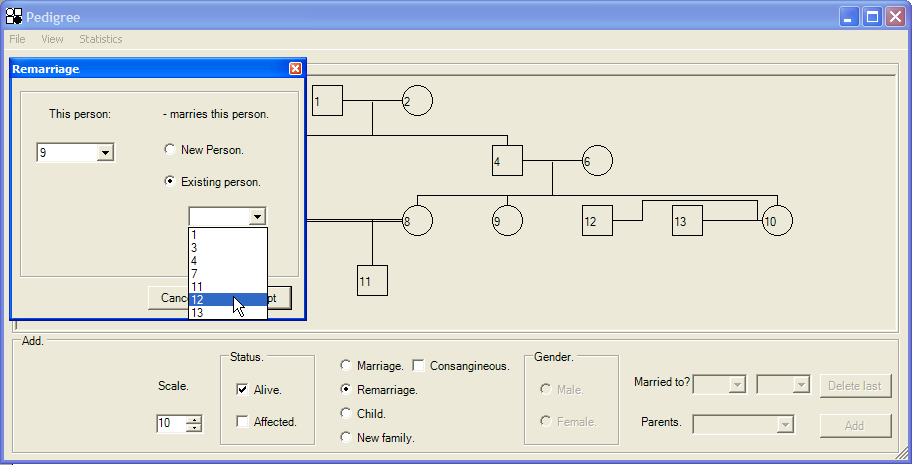
Figure 10: Addition of a second marriage between two pre-existing individuals. In this example, individual 12 is marrying his first wife’s sister, individual 9.
Figure 11 shows the pedigree created above, with the addition of children to each marriage, to illustrate where each child is drawn. Child 14 is the son of 12 (father) and 9 (mother), child 15 is the daughter of 12 (father) and 10 (mother) and 13 (husband) and 10 (wife) have a daughter 16.
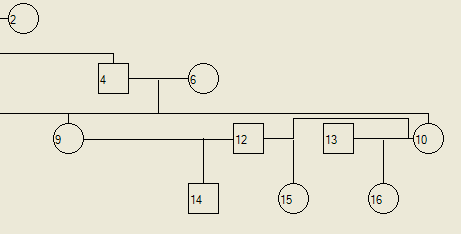
Figure 11: Positioning of children arising from remarriages within a pedigree.
It is also possible to form second marriages that are consanguineous; the program automatically detects when two individuals would produce a consanguineous union and adds double lines to signify it as a consanguineous marriage (Figure 12).
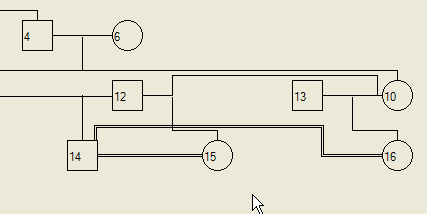
Figure 12: Remarriage between consanguineous individuals.
Unfortunately, if multiple consanguineous marriages exist within the pedigree, the lines showing the marriages may superimpose, as shown in Figure 13.
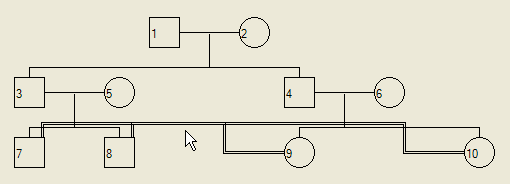
Figure 13: Example of the overlapping of two consanguineous unions, making it impossible to tell whether individual 9 is married to individual 7 or 8.
To overcome this problem, left-click on one the involved individual's symbol; this displays the Edit details dialogue box (Figure 14).
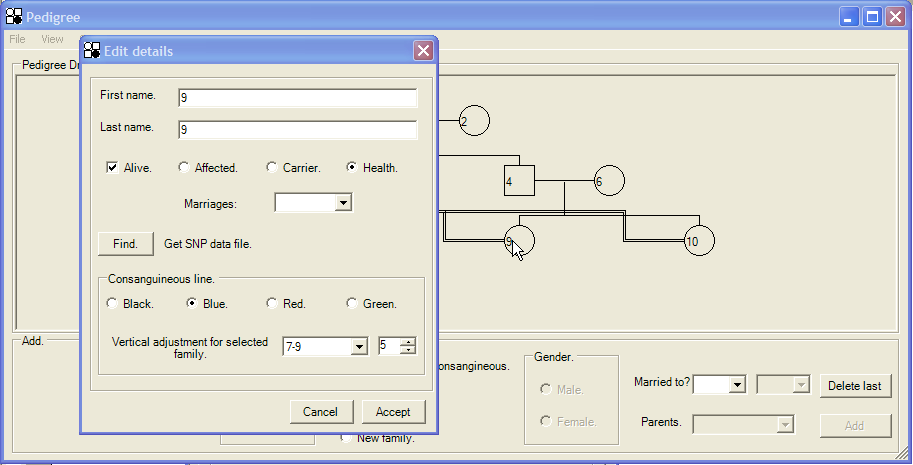
Figure 14: Edit details dialog window.
Using this dialogue, one consanguineous union may be selected from the drop-down list next to the Vertical adjustment for selected family label. You can then either change the colour of the consanguineous union lines (radio buttons) or the vertical displacement of the line (using the box to the right of the marriage selection box). Figure 15 shows the result of performing the adjustments selected in Figure 14.
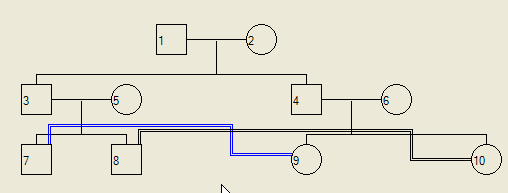
Figure 15: The display after the changes shown in Figure 14 were accepted.
2.2.3—Editing the details of individuals in a pedigree
The Edit details dialogue box can also be used to edit an individual’s details. As above, left-clicking on an individual opens this dialogue box, as in Figure 15. It is now possible to change the disease status of the individual to Healthy, Carrier or Affected, as well as assigning him or her as Alive or not. It is also possible to name the individual. (By default, the first and last names are set to the individual’s unique ID number.) The menu on the Pedigree form can be used to select display of ID numbers or names of family members (Figure 16).
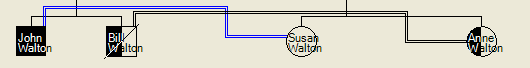
Figure 16: Affected, carrier and healthy individuals identified by their full names rather than ID numbers. Bill Walton is shown as deceased.
2.2.4—Disease status probabilities within the pedigree
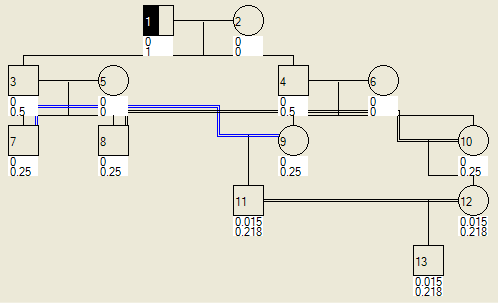
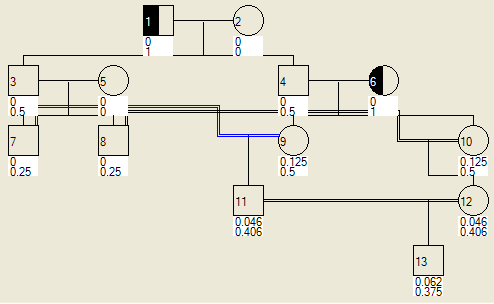
If an individual has his or her phenotype set (using the Edit details dialogue), the probability can be calculated of his or her descendants being affected or a carrier. Figure 17a shows a pedigree in which founder 1 has been assigned as a carrier, while Figure 17b shows the same pedigree with two carriers assigned.
 |
 |
| a | b |
Figure 17: Display of probabilities that individuals in the pedigree are carriers or affected, by virtue of either individual 1 (a) or individuals 1 and 6 (b) being assigned as carriers.
The upper number is the probability that the individual is affected and the lower number that he or she is a carrier. For example, in Figure 18a, individual 11 has probabilities of 0.015 of being affected and 0.218 of being a carrier, while in Figure 18b, the same individual has probabilities of 0.046 of being affected and 0.406 of being a carrier. (These figures are truncated, not rounded, so that the exact values for individual 11 in Figure 18b are in the ranges 0.04600 to 0.04699 for being affected and 0.40600 to 0.40699 for being a carrier.)
Using the menu of the Pedigree form, the view can be changed to display any of the following: no probability data, the chance that an individual is a carrier, the chance that an individual is affected, the chance that an individual is either affected or a carrier, or (as in Figures 18a and 18) both the affected and the carrier probabilities.
2.2.5—Linking Affymetrix data files to individuals
In the Edit details form, click the button. This will allow you to browse to the location of the data file and link it to the individual. If you make a mistake, you can unlink the file by clicking the button, which appears to the right of the file’s name. No data will be added to the AutoSNPa database until the Pedigree form is closed. To close the form, use its menu, to either discard all the pedigree information or add it to the database. Only if the menu option is used will the data be saved to the database. If you edit a pedigree made earlier, only the data files that you have changed will be read. Creating the database may take a couple of minutes (depending on the number of files to be read and the speed of the computer. During this time, the program will stop responding to the mouse and keyboard, but its activity can be seen as a busy hard disk.
2.2.6—Pedigrees with more than one founding family
If a pedigree contains more than one founding family, members of different families can marry by selecting them as described above. If two families intermarry a number of times, AutoSNPa can detect which marriages are consanguineous and then draw them appropriately (Figure 18). If individuals in each family are designated as affected or carriers then AutoSNPa can still calculate the probabilities as described above (Figure 19).
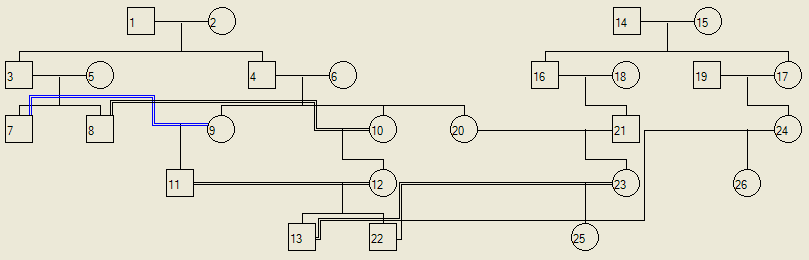
Figure 18: Two founding families (based on marriages 1-2 and 14-15) inter-marry multiple times. Marriages 21-20 and 13-24 are non-consanguineous, while the union between individuals 13 and 23 is consanguineous.
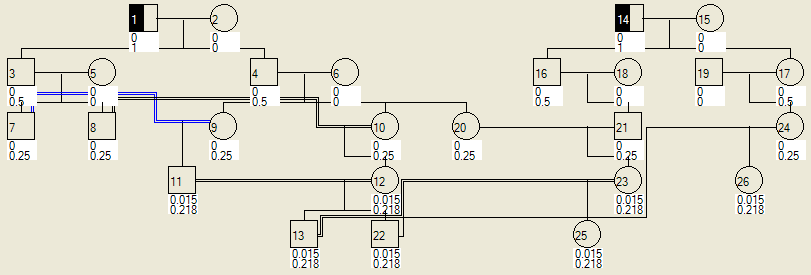
Figure 19: The chances of individuals in the two families being affected or carriers, as described in Figure 17.
2.2.7—Moving and changing the scale of the pedigree display
Families can be moved around the Pedigree window by holding down the left mouse button and dragging. The size of the pedigree can also be changed using the value in the Scale box.
2.2.8—Notes on drawing pedigrees
-
This program is primarily intended for SNP analysis, and so only marriages that contain children may be redrawn when a pedigree image is produced from a previously saved file.
-
While the program is able to draw complex pedigrees, it is possible to produce a pedigree that it is unable to draw. However, these pedigrees tend to include marriages between the children of multiple incestuous unions. In our experience, these kinds of pedigree rarely occur.
-
If the window is resized, the image may appear cropped. To see all the individuals; move the pedigree in the window as described above.
2.3. Linking data files to individuals
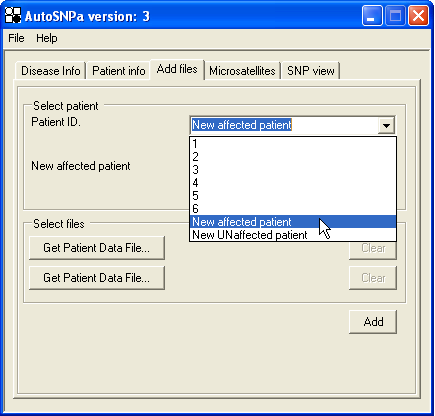
Figure 20: Affymetrix data can be linked to either people in a pedigree or new patients.
This refers to the third tab Add files on the startup window (Figure 20). Affymetrix SNP data files can be linked either to individuals in a pedigree or to a new generic patient, affected or unaffected . To enter data files, first select a patient from the Patient ID list and then press one of the buttons. If you need to link two different data files to an individual (e.g. a Sty and a Nsp file), use both buttons; the file name will then be shown next to the button. The order in which the buttons are used is not important. If the or option is selected, a new individual will be added to the system. If a second data file is linked to a pre-existing individual link the genotype data for the first file will be deleted and re-entered along with the new second file's genotype data. Since the program must re-access the first file, it must be either in the original location or reselected. Finally, to add the files, press the button.
2.4—Adding microsatellite genotype data
Rather than generating Affymetrix whole-genome SNP data for all of members of a pedigree, it may sometimes be desirable to analyse a subset of patients for homozygous regions and then study the remaining individuals with targetted microsatellites to confirm or refine the presence of a homozygous region. To incorporate such an approach, the current version of AutoSNPa allows the combining of microsatellite genotypes with SNP data.
2.4.1—Adding microsatellite markers
This is done by entering the microsatellite name and positional data in the text-boxes highlighted by the red ellipse in Figure 21. Only physical positional data are mandatory,while genetic positional information may be omitted. The microsatellites are viewed one chromosome at a time; thus, only microsatellites located on the chromosome selected using the Chromosome list box (left most box in the red and blue ellipse) are shown. Microsatellites are added by pressing the button and can be either deleted or updated using the and buttons. (If a microsatellite is deleted, all genotype data linked to it will also be lost.)
2.4.2—Entering microsatellite genotypes
Patient genotypes are added via the text-boxes in the blue ellipse in Figure 21. To add a genotype, first select a chromosome and then pick a microsatellite marker from the list box labelled Name. Next, select a patient from the Patient ID list (which only includes individuals with Affymetrix data files linked to them). If no Affymetrix data is available for a patient, link the individual to the “blank” data file that is included in the AutoSNPa download bundle. This file only contains data on the two SNPs per chromosome which reside at the p and q arm telomeres. Once the microsatellite and patient have both been selected, enter the genotypes into the Genotype text box. If the genotype is homozygous, enter the allele size in base pairs as a single digit (i.e. ‘100’ or ‘254’). If heterozygous, enter both allele sizes separated by a ‘/’ (i.e. ‘100/254’ or ‘200/254’). To delete a genotype, enter ‘--/--’ into the Genotype text box. Pressing the button below the Genotype text box will now update the genotype data.
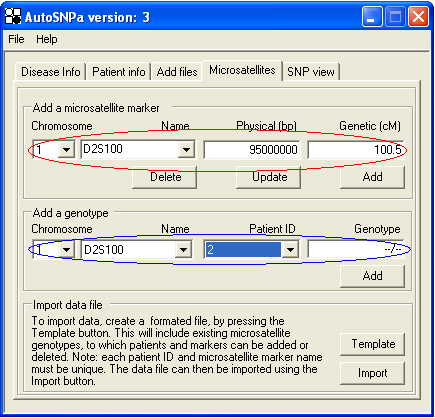
Figure 21: Adding microsatellite genotype data using the Microsatellite tab.
2.4.3—Exporting and importing microsatellite data
AutoSNPa can both import and export microsatellite data. However, the files are specific to the single pedigree under analysis. To export microsatellite data, press the button, which will create a tab-delimited text file with a *.GEN file extension. This file contains information on the individuals and microsatellites in the database and their genotypes (Table 2). Each line in the file begins with either a “:”, “~” or “£” symbol, with the following meanings: “:” signifies patient information; these lines display the patient ID, father's ID, mother's ID, sex, first name, last name, status (True = affected) and Affymetrix file location. The line starting with “~” displays the patient IDs for all patients with genotype data. Finally the lines starting with a “£” contain the microsatellite information (marker name, chromosome, physical position (bp) and genetic position (cM)) followed by the genotypes. The genotypes are given in the same order as the patient IDs in the line starting with the “~” symbol. The size of each allele is stated, and where no data is available the genotype is declared as “-1/-1”. The patient information is ignored when importing data and is included only to help identify each patient's ID number.
Since the exported template file consists of simple tab-delimited text, further microsatellites and genotype information can be added using a text editor. However, data cannot be added for patients with no SNP data. Provided the genotypes are correctly formatted to match the patient list, this allows effective batch-wise importing of microsatellite data using the button. Note that when microsatellite data are imported, all existing microsatellite data are overwritten.
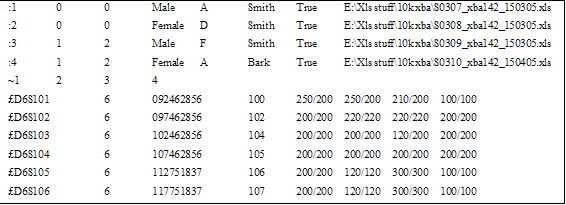
Table 2: Structure of a microsatellite import file.
2.4.4—Saving the data to file
Once all the SNP and microsatellite genotypes and patient data have been entered, the information can be saved to disk via the menu (Figure 22). The data is saved as a tab-delimited text file, but in this case with a *.SNP file extension.
Figure 22: Options for opening and saving the SNP database.
3—Analysis of SNP data
Once the SNP database has been constructed, it is time to view and analyse the SNP data using AutoSNPa. This is done through the fifth and final tab on the startup window, SNP view.
3.1—Viewing the SNP data
By clicking the button (Figure 23), the Chromosome View window is opened for display of the SNP data (Figure 24).
Figure 23: SNP data can be visualized by clicking the Viewbutton.
In Chromosome View, each chromosome is displayed separately, and accessed by clicking on its tab at the top of the SNP data image (Figure 24). As the cursor is moved over the genotype data, the individual’s name is displayed alongside the chromosome number. (If the individual’s name has not been specified, then their unique ID number will be shown instead.) and the map location of the cursor is shown below its icon.
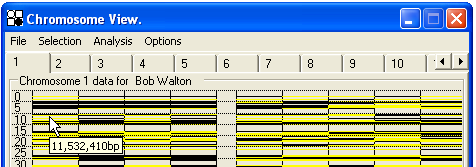
Figure 24: Each chromosome is selected by clicking the appropriate tab (In this figure the cursor has selected Chromosome 1.)
Initially, the genotyping data are shown as an array of colour-coded SNPs, placed along the chromosome ideogram according to physical map position (Figure 24-25). By selecting from the menu , the genotyping data can instead be displayed relative to genetic position (Marshfield averaged distance) (Figure 26). A comparison of Figures 25 and 26 demonstrates the difference this can make, as the regions around the centromere and rRNA clusters are shortened and the telomeric regions expanded when viewed by genetic distance.
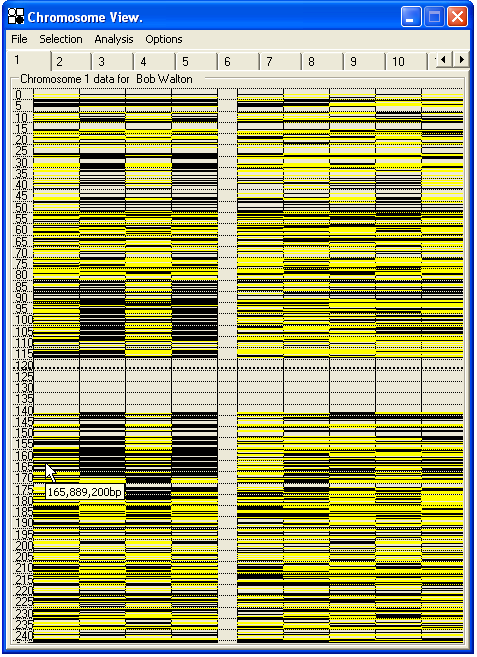
Figure 25: SNP data for Chromosome 1, arranged along the chromosome by physical position; the numbers to the left show the distance from the p arm telomere in Mb.
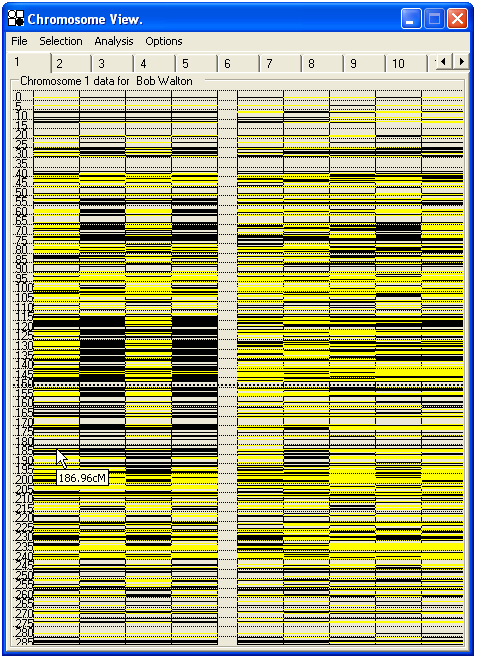
Figure 26: SNP data for Chromosome 1 displayed by genetic position; the numbers to the left of the SNP data show the map distance in cM.
In these displays, each individual's data occupies a single column, with the columns grouped according to “affected” and “unaffected” disease status; affected individuals' data is displayed in the left of the two groups. Each SNP genotype is colour-coded, depending on its categorization as “no-call”, “heterozygous”, “common homozygous” or “rare homozygous”. (If both AA and BB genotypes for a SNP are present in different affected individuals, the variant that is most common is designated the “common homozygous” genotype; genotypes of unaffected individuals are not used in arriving at this decision.) The colour assigned to each category can be set by the user (Figure 27). However, by default, the “common homozygous” category is coloured black and the “rare homozygous” and “heterozygous” genotypes are both yellow. This allows homozygous regions common to all patients to be identified by the absence of yellow markers.
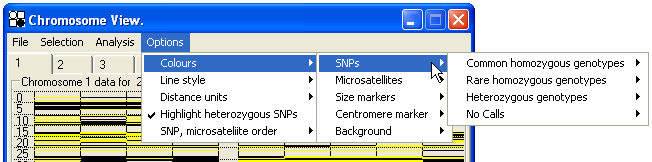
Figure 27: The genotype colour code can be changed via the Options > colours menu for both SNP and microsatellites.
Smaller regions of this display can be mouse-selected by placing the cursor at the top of the region, and holding down the left mouse button while dragging down to the bottom of the region, before releasing the mouse button. This will draw a red box around the selected region, which can be expanded for easier viewing by pressing the Enter key or choosing from the menu. (Figure 28). The expanded region can be saved by clicking the menu option, and then reviewed by clicking the menu (Figure 29). The selected region will also be saved in the *.SNP data file and so can be re-examined when the data are later re-opened in AutoSNPa.
 |
 |
| a | b |
Figure 28: The Chromosome View can be expanded by selecting a region with the mouse (a) and then either selecting Draw from the Selection menu or pressing the Enter key. If the Add menu option is then clicked, the selection will be saved as a sub-menu under the main Selection (b).
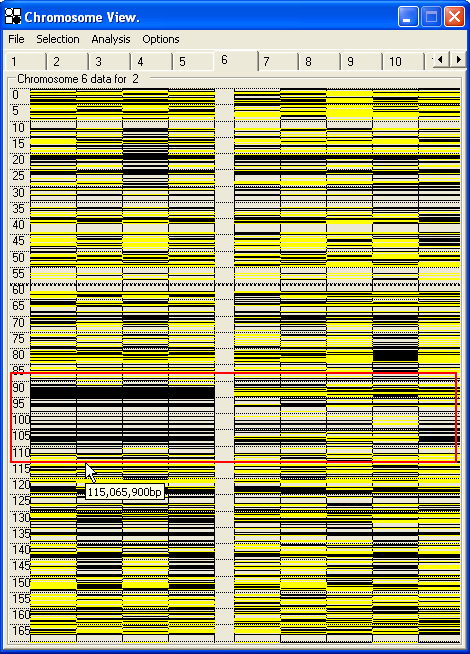
The expanded region can then be moved along the chromosome using the Up and Down arrow keys; each view overlaps the previous one by 10% of the region's length. To view the whole chromosome again, press the Esc key. Clicking the right mouse button at any position will identify those SNPs lying in the vicinity of the selected point (Figure 29).
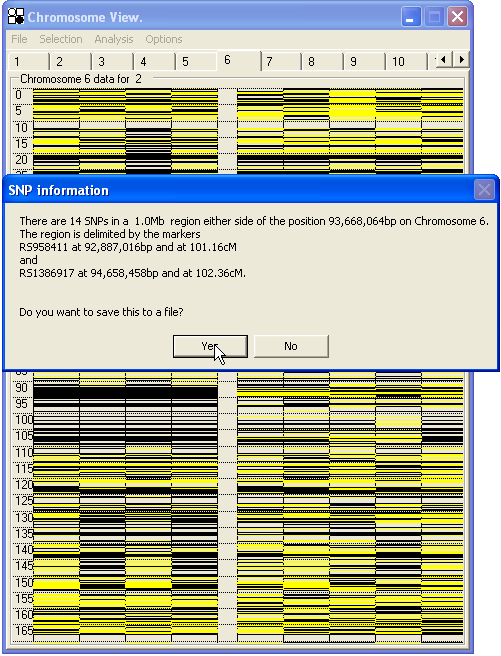
Figure 29: Right-clicking on the form generates a dialogue box showing details of the markers lying around the cursor. If the region contains more than 14 SNPs, only the upper and lower SNP will be identified on screen; however if the data are saved to file, all SNPs will be shown.
3.1.1—A note on the display method
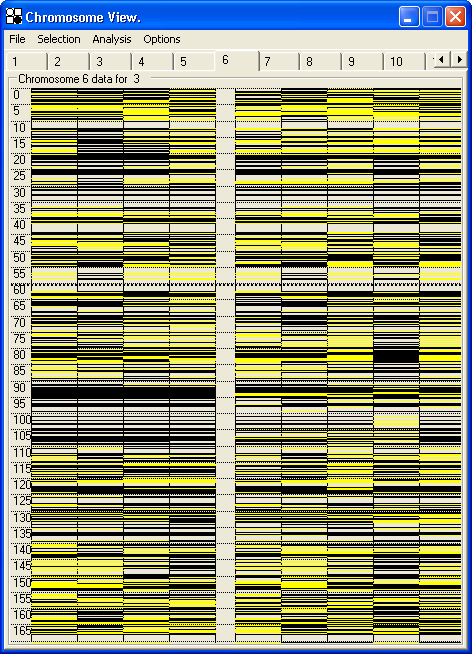
Since the number of SNPs present on each chromosome is likely to vastly exceed the vertical screen resolution, many conflicting SNP genotypes are likely to map to the same display pixel. Since in an autozygosity mapping project, it is the heterozygous genotypes which define the region of interest, AutoSNPa gives special emphasis to heterozygous SNP markers. To highlight their presence, SNP the chromosome view is drawn twice in succession; the first pass includes all the genotypes, while the second (overwriting) pass only includes heterozygous SNPs. While this increases the time taken to display the data, it has the beneficial effect of highlighting more effectively the regions of homozygosity (Figure 30a and 30b).
 |
 |
| a | b |
Figure 30: Shows the effect of turning on (a) or off (b) the option, which performs an overlay of heterozygous (in this case yellow) SNPs, so as more effectively to highlight genuinely homozygous regions.
3.1.2—Different methods for viewing SNP data
As mentioned above, precise depiction of all SNP data relative to aphysical or genetic map is limited by screen resolution. To alleviate the masking of genetic information at multiple superimposed SNP loci, AutoSNPa can generate three different graphical representations of the SNP data, as well as a text file detailing any region of interest. Each of the three graphics represents the data in a different way from the initial chromosome display, but uses the same y axis scale and distance units (cM or Mb), so that both views can be directly compared. All of these functions are accessed via the menu (Figure 31). The menu also allows you to select a number of additional non-visual functions.
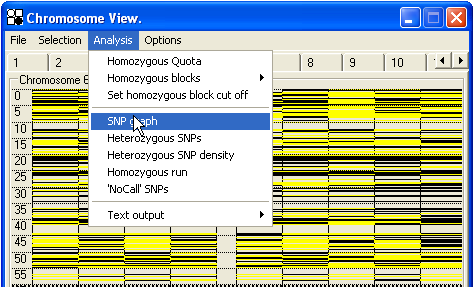
Figure 31: The Analysis menu.
3.1.3—Visual analysis options for SNP genotype data
3.1.3.1—SNP graph
In this view, all the SNPs that lie superimposed at each y axis pixel are shown as a colour-coded horizontal bar, using the same default colours as described above (Figure 32). Affected individuals are again shown in a panel to the left of the unaffected individuals. Genuinely homozygous regions then appear as black bars, whereas regions that harbour “rare homozygous” and heterozygous genotypes contain a black bar tipped by a yellow end. (“No-calls” are shown as white bars). As with the SNP graph, the underlying SNP data can be interrogated via the mouse buttons.
This view is useful for revealing heterozygous SNPs that lie very close to homozygous SNP runs, since some SNPs may be separated by less than 200bp. Because of the greater density of information, it is especially useful when viewing higher density (50k-1M) SNP data.
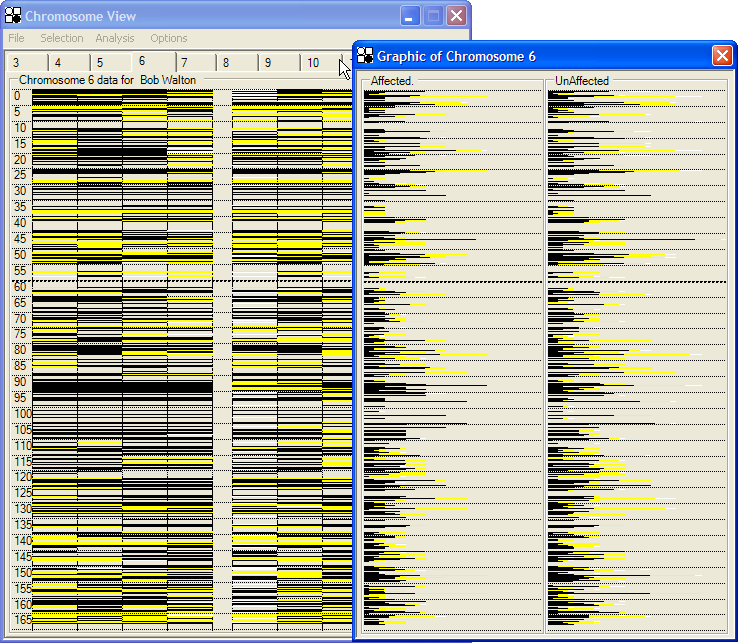
Figure 32: SNP graph view of the genotype data for chromosome 6.
3.1.3.2—Heterozygous SNPs
This view is very similar to the initial chromosome view, but only heterozygous SNPs are shown, enabling rapid identification of homozygous regions (Figure 33c). Extended regions that are homozygous for the same SNP genotype in all affected patients are highlighted by a pale blue box, whereas regions homozygous for different genotypes are shown in pink (Figure 33b). These features can be turned on/off via the menu. The preset default is to highlight runs longer than five SNPs, but this cut-off value can be changed to suit each pedigree via the Set homozygous block cut off option (Figure 31).
For a detailed view of the underlying genotype data, clicking the mouse buttons produces either a colour-coded Excel spreadsheet or a tab-delimited text file organised in columns, showing the SNPs’ IDs, physical map positions and genotypes, for each individual. This output file also contains the URLs and search strings for the Ensembl and UCSC Golden Path genome browsers, allowing the rapid identification of genes within this chromosomal region. Clicking the left mouse button displays data for the minimum homozygous region (red bar in Figure 33a), whereas the right mouse button retrieves data for a region delimited by the longest runs of homozygous SNPs above and below the selected region (green bar in Figure 33a). This latter feature is designed to overcome the possible artefactual truncation of a homozygous region by a wrongly-called SNP. Examples of the files produced can be seen in the Appendix.
 a |
 c |
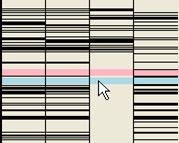 b |
Figure 33: Heterozygous SNPs view. In a, the green line illustrates the extent of the SNP data saved to file if the right mouse button is pressed when the cursor is over the blue box, and the red line shows the extent of the SNP data returned if the left mouse button is activated. Pale blue shading highlights regions where all affected patients share the same genotype; pink regions are where all patients are homozygous, but for any allele.
3.1.3.3—Heterozygous SNP density
This view was designed to aid the user in dealing with data sets that have a high level of false heterozygous SNP calls. This can be an important problem when trying to define the extent of an autozygous region; false heterozygous calls may lead to a region being rejected from consideration, when in fact it lies within an autozygous region.
Rather than displaying each SNP genotype individually, the view displays the data as a grey-scale image, in which regions with few heterozygous SNPs are shown as black, while regions with more heterozygous SNPs are white. Typically, 70% of SNPs are homozygous even in a non-autozygous region. Therefore, the scaling of grey shades is set so that pure white is displayed when 66% of the SNPs in a region are heterozygous. (Positions that do not contain any SNPs have the same colour as the window background.)

Figure 34: Heterozygous SNP density view
3.1.3.4—Homozygous runs
In this view, runs of homozygous SNPs are displayed as continuous lines (Figure 35a, 35b). The displacement of the line along the x axis depends on a simple estimate of the statistical significance of a run of this length. The lower the probability that such a run represents a random event, the farther to the right the line is drawn; a line to the extreme right has a nominal probability of <10-4 of being a chance occurrence. This probability can be calculated either from the proportion of homozygous SNPs on that individual’s chromosome (Figure 35a) or from the allele frequencies supplied by Affymetrix (Figure 35b). We emphasize that neither of these estimates is biologically robust, both because of the unknown true allele frequencies and because of unknown degrees of linkage disequilibrium within marker runs. Rather, the intention is to give a qualitative indication of which homozygous runs may be chance occurrences, and which may be due to inheritance from a common ancestor. As described above (see ), runs of homozygous SNPs common to all affected individuals are shown by blue or pink rectangles, and the underlying SNP data can be accessed via the mouse buttons. This allows the user to make rapid judgements about the interest of a region, taking into account factors such as the number and density of homozygous SNPs.
 |
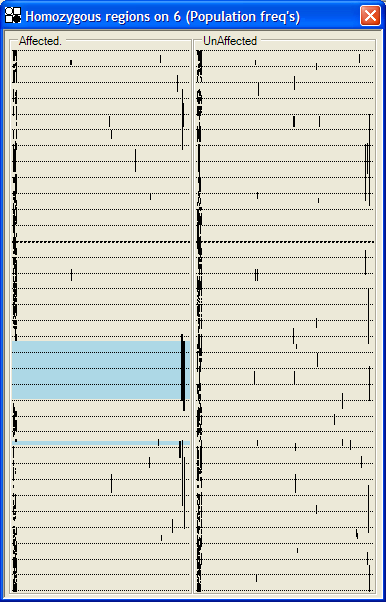 |
| a | b |
Figure 35: Homozygous runs views, in which each vertical line shows the length of a homozygous region, while its displacement to the right indicates the nominal probability that the region is the result of chance. This nominal probability can be calculated using either the proportion of homozygous SNPs that a patient has on the chromosome under analysis (a) or from the predetermined population allele frequencies (b).
3.1.3.5—NoCall SNPs
This view is very similar to the view, but instead of showing the heterozygous SNPs, only SNPs that produced a ‘NoCall’ result are shown. This allows the identification of datasets or regions that contain high levels of ambiguous SNP calls. This may be useful, for example, in the case of regions that are involved in duplications and deletions, where ambiguous fluoresence signals on the SNP chip result from aneuploid gene dosage (Figure 36).
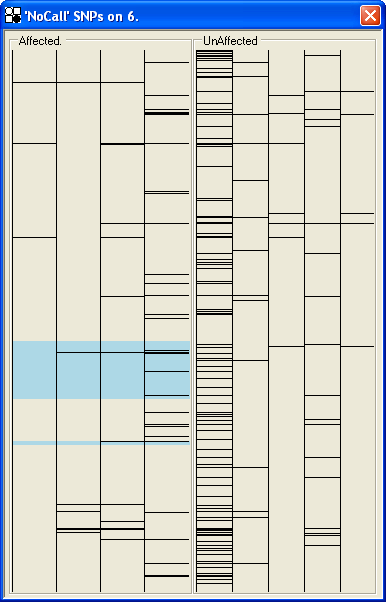
Figure 36: NoCall SNPs view; each horizontal line identifies the position of a NoCall SNP result. The first unaffected individual shows a comparatively high NoCall rate.
3.1.4—Text-based options for analysis of SNP genotype data
3.1.4.1—Homozygous blocks
These functions are accessed via the menu (Figure 31) and generate text files that detail each homozygous region of interest (those shown as blue or pink rectangles in the graphical views described above). These can be either flat text files or colour-coded Excel spreadsheets. The former option makes it easier to include the data in a report, while the latter enables regions of SNP genotypes common to the affected individuals and an unaffected individual to be rapidly identified.
There are three different homozygous block file output options, , and . The and options produce text files which contains the data equivalent to the blue and pink rectangles, respectively. The option allows you to place affected individuals into hereditary units which represent different pedigrees. Each unit is then analysed individually for homozygous runs of the same genotype across all individuals within that unit. The results for all of the units are then compared, and regions that have homozygous regions across all units are written to the file.
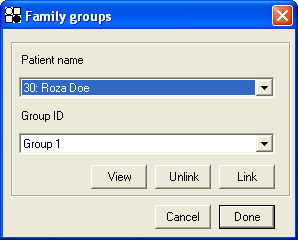
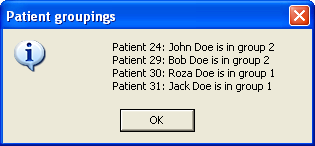
These groups or units are created using the window shown in Figure 37a. Each individual is selected from the top drop-down list, and then linked to a group, chosen from the lower list, by pressing the button. Patients can then be unlinked, either by placing them in a different group, or by selecting their ID/name and clicking the button. The current groupings can be seen via the button, which creates a message box listing the patients and their grouping (Figure 37b). The output file is then produced by pressing . When using the option, any patients not explicitly linked into a group are disregarded when selecting regions for export. This allows individual patients with suspect SNP data to be arbitrarily excluded from the analysis.
 |
 |
| a | b |
Figure 37: The Family groups window (a) allows you to place individuals into different hereditary units or groupings, while the Patient groupings window (b) shows information on the current patient groupings.
If the menu option is checked, the program cycles through each chromosome in turn, allowing them to be rapidly analysed. If this menu is checked and the analysis function is chosen, the selected data from all the chromosomes are placed in the resultant text file. The analysis option calculates the number of homozygous SNPs on the current chromosome, for each individual. This gives an indication of the degree of inbreeding that each individual has for the current chromosome. This may be used to inform the relative importance assigned to homozygous runs on that chromosome.
4—Computer hardware and software requirements
AutoSNPa should run on any computer that has the Microsoft .NET framework version 2 or later; we have tested it on Windows XP SP3, Vista SP1 and Windows 7 RC. The .NET framework is available free from Microsoft (search www.microsoft.com for “Microsoft .NET Framework Version 2 Redistributable Package” or install via Windows Update). There is no minimum hardware specification for AutoSNPa, other than that needed to run the .NET framework, but obviously, the slower the CPU and smaller the RAM, the slower the program will run. The forms produced by the program cannot be resized, and so to view them the screen must have a vertical resolution greater than 600 pixels.
AutoSNPa does not have intrinsic limitations on the size of data files, and can load and display data from 50k, 250k, SNP5 and SNP6 arrays effectively. However, with large datasets, hardware limitations need to be considered. If the overall size of the SNP database exceeds available RAM, the program will (like any other) slow down as it swaps information to disk.
AutoSNPa can run on any computer with the .NET Framework, even without Administrator rights to install software (e.g. a computer in a departmental cluster).