Quick user guide with the ATOH7 dataset
Introduction
This guide is a quick demonstration of the use of AgileVariantViewer with data used to identify a mutation in the ATOH7. Since this analysis identified a number of possible deleterious sequence variants it was necessary to screen the data using AgileGeneFilterer it prioritise the screening of the patients sequence variants in the patients. As with the PXDN data, this data is derived from a single sequencing run using a DNA sample enriched for a number of genomic regions.
Entering the sequence variants data
Enter the the gemomic annotation file and the read depth file created by AgileAnnotator and the sequence variants file created by AgileAnnotator and optionally filtered by AgileGeneFilterer.
File formats
A description of the file formats for the variant and read depth files used by AgileVariantViewer can be found here.
Viewing the sequence variant data
When the Data view window opens it displays a graphical view of the analysis data for chromosome 1 in the upper panel (Figure 2). Below the graphical display panel are five other panels which allow the sequence variants to be filtered and then exported. Each of these panels is described in detail below:
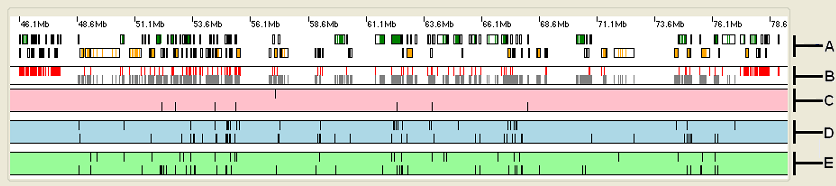
Figure 3: The upper panel displays the analysis data organised as a number of horizontal strips.
The upper panel displays the analysis data organised as five horizontal strips (Figure 3).
- Strip A: shows the location of any genes (black rectangles) in the selected region with the green and orange rectangles representing exons transcribed from
the positive and negative DNA strands respectively.
Placing the cursor over a gene in this strip causes the gene's name to appear in the windows . - Strip B: shows the position of exons whose typical read depth is above (grey block) or below (red block) the cut off value selected from the list in the Region view options panel (see below).
- Strip C: shows the location of sequence variants that will affect function of a protein. These variants include those that create in frame stop codons, insertions and deletions and splice site variants. The vertical lines at the top of the strip represent homozygous variants, while the vertical lines at the bottom of the strip correspond to heterozygous variants.
- Strip D: shows variants that change the amino acid sequence of a protein as well as variants close to the start codon which may affect protein translation, these variants may or may not affect the proteins function. The vertical lines at the top of the strip represent homozygous variants, while the vertical lines at the bottom of the strip correspond to heterozygous variants.
- Strip E: shows sequence variants that are unlikely to affect a protein's function since they are either in an intron or do not change a proteins amino acid sequence. Again, the vertical lines at the top of the strip represent homozygous variants, while the vertical lines at the bottom of the strip correspond to heterozygous variants.
Select the candidate region if known
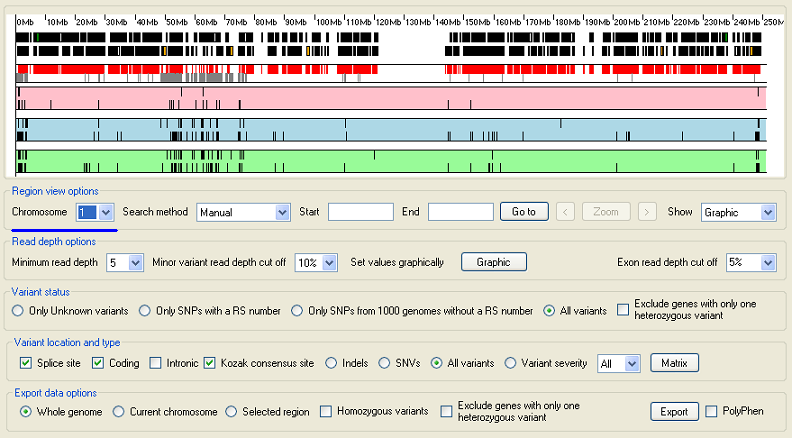
Figure 2: Selecting a chromosome.
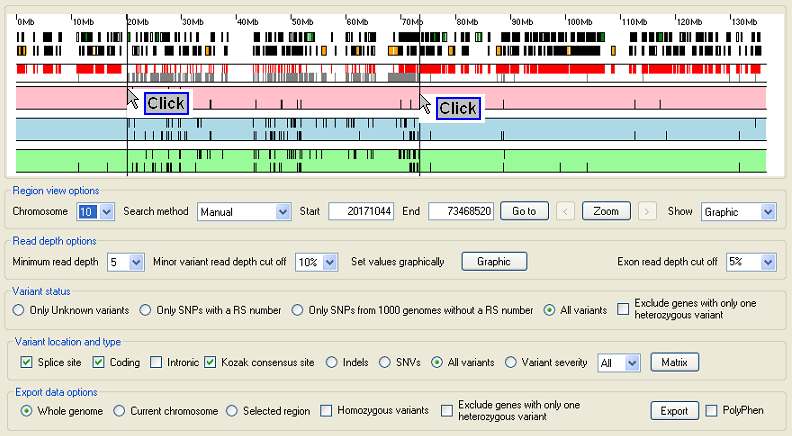
Figure 3: Select a specific region.
To select a sub region set the list in the Region view options panel to manual and place the cursor over the end of the region and press the right hand mouse button. Then place the cursor over the start of the region and press the lefthand mouse button. This should place two vertical lines that delimit the region of interest (Figure 3), to zoom in to the region press the button in the Region view options panel (Figure 4).
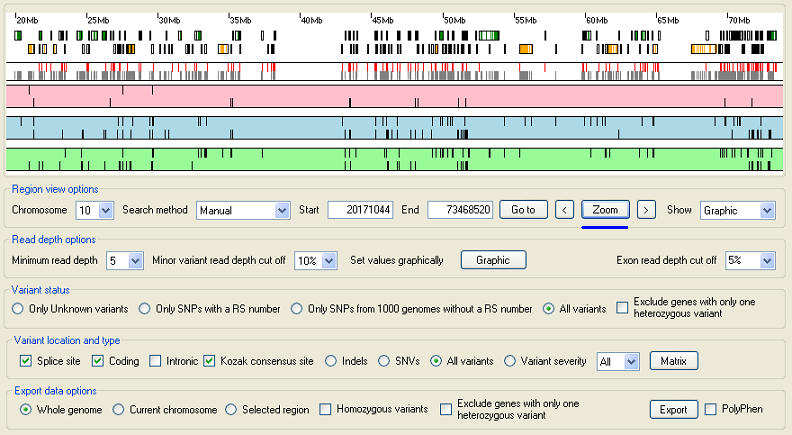
Figure 4: Zoom in to a region by pressing the button.
Adjust the variant cut off parameters.
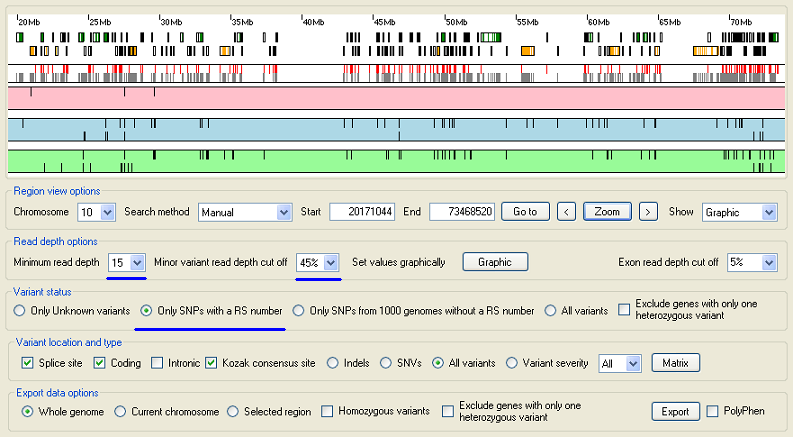
Figure 5: Adjust the cut off parameters.
The sequence variant data used in this example has been filtered using AgileGeneFilterer so those variants with a RS number can be identified. Since sequence variants with a RS number are probably true positives use this to set the cut off values which discord false positives, but retain true positives. Select the option in the Variant status panel. Then increase the and values in the Read depth options panel until sequence variants start to disappear, but the majority of sequence variants are still visible. If the region is autozygous, try to remove all of the heterozygous variants. However some variants that appear to be heterozygous may remain if the region is duplicated elsewhere in the genome. If sequence reads derived from the duplicated sequences are slightly divergent, but still mapped to the region of interest the divergent positions will appear as heterozygous variants.
Viewing sequence variants not found in the 1000 Genome Project
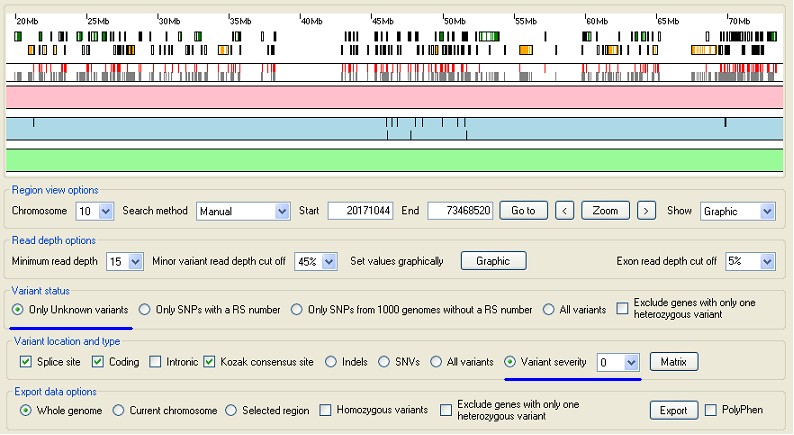
Figure 6: Viewing sequence variants not found by the 1000 Genome Project.
Select the option in the Variant status panel to display sequence variants that have not been identified in the 1000 Genome Project. If the region is autozygous, the majority of the variants should be homozygous. If a large proportion of the variants are heterozygous, increase the and values in the Read depth options panel until the majority of variants are homozygous. Select the option in the Variant location and type panel and chane the value in the list to 0 to discord all variants that do not affect a protein's amino acid sequence.
Exporting sequence variants
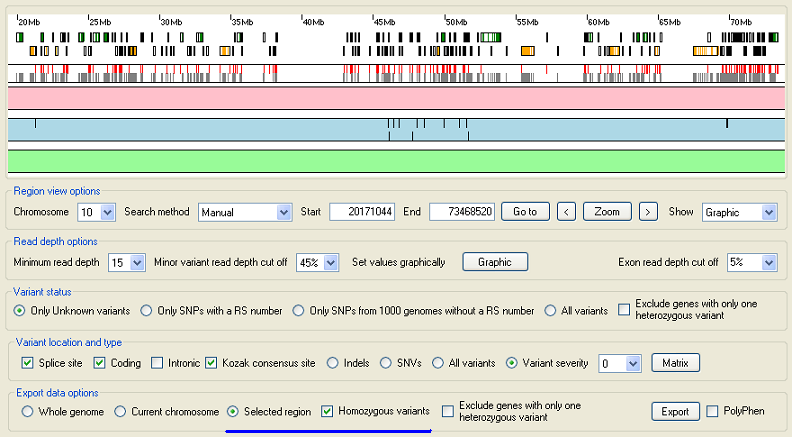
Figure 7: Exporting sequence variant data