User guide
Introduction
AgileSAMFileSorter sorts unordered *.sam files by creating a series of smaller files, each of which contains a sorted subset of the original reads. These files are then merged into a single sorted *.sam file, whose name is the original file’s name with “_ordered” appended to it (e.g. ATOH7.sam → ATOH7_ordered.sam).
Warning: During the ordering process, the folder may contain a copy of the original file, all the sorted fragment files and a copy of the sorted *.sam file. Consequently, it is important that there is plenty of free disk space during sorting. Also, since AgileSAMFileSorter reads and writes a large amount of data, the data files should be on a local hard disk. If the original unsorted *.sam file is on a network drive or a USB stick, the sorting process may take a VERY long time to complete.
Sorting a SAM file.
To sort an unsorted *.sam file, press the button, choose the file, and then press . If the sorted *.sam file is to be used on a Linux or Apple computer select the line break option (Figure 1). The sorting process is performed in six steps, as outlined below:
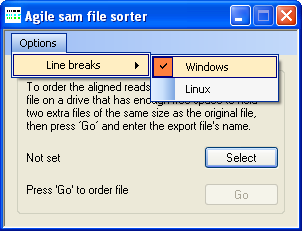
Figure 1: The option creates files with Linux compatible line breaks
- Read the unsorted *.sam until 2,000,000 correctly formatted and aligned sequence reads have been found.
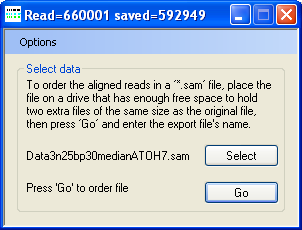
Figure 2: During the data reading phase, AgileSAMFileSorter displays in its title bar the number of sequences read and the number it has saved.
- Sort the 2,000,000 reads by chromosome and chromosomal position.
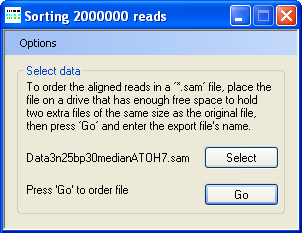
Figure 3: When sorting the aligned sequences, AgileSAMFileSorter displays “Sorting 2000000 reads” in its title bar.
- Save the sorted reads to a fragment file.
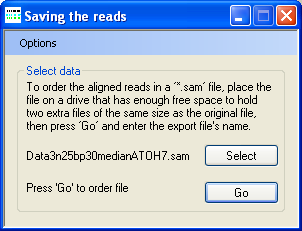
Figure 4: When saving the sorted aligned sequences, AgileSAMFileSorter displays “Saving the reads” in its title bar.
- If the entire unsorted *.sam file has been read, go to Step 5, otherwise go to Step 1 and repeat the process.
- Merge the reads in the sorted fragments, to create a single sorted *.sam file.
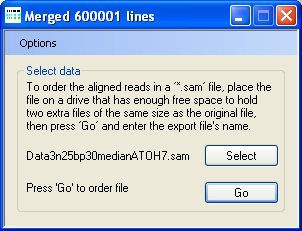
Figure 5: When merging the contents of the sorted fragment files into a single file, the AgileSAMFileSorter title bar displays the number of reads exported to the new sorted *.sam file.
- When all of the fragment files have been read, AgileSAMFileSorter will attempt to delete the fragment files. Depending on the security settings of the operating system, this may sometimes fail. If any fragment files do remain after the sorting process, it is very important that they are deleted before sorting another *.sam file in the same folder.
During the final merging phase, the working directory may occupy three times the space of the original unsorted *.sam file. It is therefore important to sort data on a hard disk with plenty of free space.
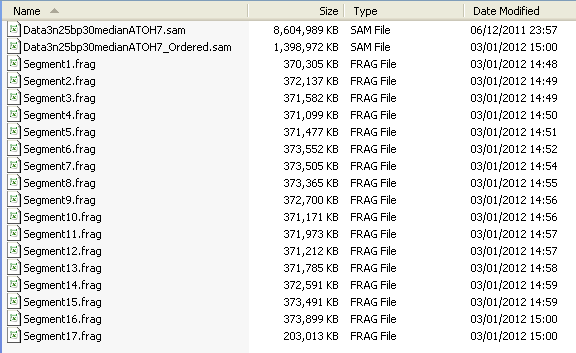
Figure 6: During the sorting process a large amount of data is stored in the folder.