User guide
Introduction
AgileSMPoint identifies somatic sequence variants occurring at specific positions in unaligned clonal (“next-generation”) sequence data. Somatic mutations of specific nucleotides cause (or play a role in the progression of) many diseases, but most notably many cancers. For instance, mutations at codons 12 and 13 in KRAS are a reproducible finding in many tumour types. The detection of somatic mutations is hindered by the presence of varying amounts of normal tissue in the DNA sample. Consequently, important somatic mutations may occur at levels of only 1 or 2% in the analysed DNA sample. To aid the detection of somatic mutations, we have developed AgileSMPoint a program which screens next-generation sequence data derived from PCR amplicons spanning the mutation hotspots of interest. AgileSMPoint does not require the sequence data to be prealigned, and is able to import from either *.fasta or *.fastq files. The sequence data to be analysed may include reads from a large number of different amplicons, each containing a number of different hotspots.
If a PCR amplicon is longer than a typical read length, it is possible to sequence it using paired ends and then combine the read data into a single full length read using AgilePairedEndReadsCombiner.
Importing sequence read data and somatic mutation hotspot location data
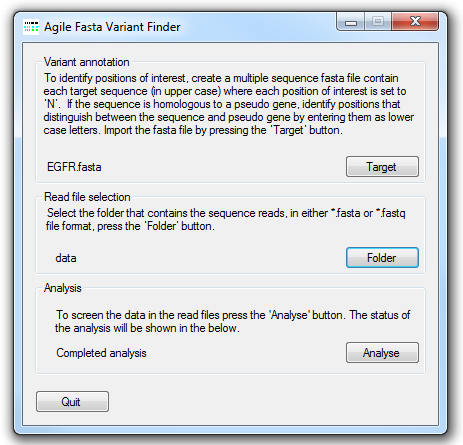
Figure 1: AgileSMPoint user interface
The file specifying the locations of somatic mutational hotspots of interest is selected by clicking the button and navigating to the target file. The structure of this file is described below. Next, select the folder containing the sequence read data (in either *.fasta or *.fastq file format) by clicking the button. Each data file may contain data from a number of different amplicons, but each amplicon should have been amplified from a single sample. The analysis is started by pressing the button and entering the name of the export file. AgileSMPoint will read and analyse the data in each file in turn and export the results to two results files. One of these contains the mutation report and the other the raw data used to create the report.
Input file formats
AgileSMPoint can import sequence data from either *.fasta or *.fastq files. The information specifying the locations of the somatic mutational hotspots is imported from a FASTA-like file, as described below:
File specifying the somatic mutation hotspots
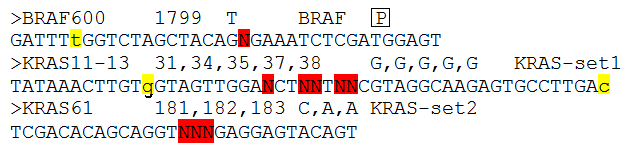
Figure 2: Each target has a description line and a sequence line
Sequence line:
This line shows the sequence spanning the positions that are to be analysed (positions of interest). The positions of interest are identified by being written as upper-case 'N's (highlighted in red in Fig. 2). Where the possibility exists that sequence reads may originate from a related homologous sequence (such as a pseudogene), such reads can be selectively ignored, provided the pseudogene differs at one or more nucleotides close to the query positions of interest. To mark a nucleotide as one serving to distinguish between pseudogene and the desired target gene, the desired gene sequence should be displayed in lower case at the divergent position (highlighted in yellow in Fig. 2). The rest of the sequence must be upper case, and include ≥20 nt both 5′ to the first 'N' and 3′ to the last 'N'.
Description line:
This line starts with a '>' character and then has four required fields. The first is just a label that identifies that particular set of results from the others in the exported data file. Then, after a tab character is a list of the positions of interest, as they are defined within a reference file (typically a cDNA sequence). There should be one number for each 'N' in the sequence, each separated by a comma. These coordinates are only used by the program when it is annotating the results for export in the output file.
Following another tab character is a list of the reference nucleotides at the positions of interest, again one character corresponding to each 'N' in the sequence line, comma-separated.
After another tab character is a 'SET' name. This is used to group target sequences that are close enough to each other to occur on the same read. Normally, if a read is found to match a target, the program does not attempt to match it to any other target, since targets are assumed to be mutually exclusive. However, if two targets are close enough that it is possible for one sequence read to contain information on both targets, the program will attempt to match a read to all the targets in a 'Set' (i.e. having the same 'Set' name), irrespective of whether that read has already been matched to a target in the same 'Set'. Typically, a set of mutational hotspots should be separated across different targets if the distance between the first and last 'N' in the sequence is greater than 30 bp. If this is not done, as the distance between the 'N's becomes too large, it becomes increasingly difficult to map all the positions, due to the requirement for a perfect match on both sides of the variant region.
As mentioned above, a read’s origin is identified using the sequences flanking the 'N' positions; if these regions are known to contains a polymorphic position, an optional field containing the upper-case character 'P' should be added to the Description line (highlighted by a black square in the first line of Figure 2). In the presence of this field, AgileSMPoint will allow for one polymorphic base in the flanking sequence either side of the 'N's.
Exported data file formats
Report file
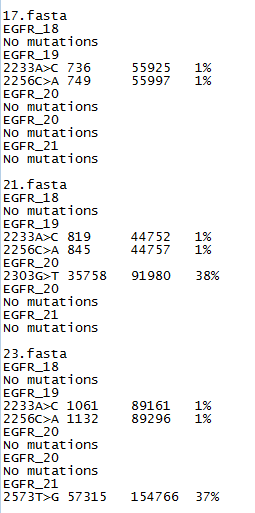
Figure 3: The somatic mutation data file format
The exported data is saved to two files, one containing a variant report and the second containing the raw data for each of the mutational hotspots. The report file lists those positions at which more than 1% of the reads contain a non-reference nucleotide (Figure 3). A variant may be annotated with an 'N' (e.g. 223A>N); this suggests that a high number of uncalled bases was found at this position. This variant could be ignored or the data prefiltered using AgileQualityFilter to reduce the proportion of low-quality reads. If the 'N' variants persist or are large in number, the sequencing quality may be poor and merit resequencing.
The report file lists the variants first by the name of the sequence read data file, and then by each target’s name. If no variants are found, the export data contains the phrase 'No Mutations'. If variants are found, then each is annotated using the reference number given in the 'Description line' of the target file (Figure 2). The annotation specifies the number of variant reads, the total number of reads and the percentage of variant reads. If a read appears to contain an indel, AgileSMPoint will note this possible indel’s position and sequence, and then if more than 1% of reads are found to contain the same indel, it is annotated and exported in the results file. Reads with indels are not used to detect single-base somatic mutations; even so, samples with an indel are also associated with a number of false positive variants that occur at a low frequency. An example of this type of output file can be found here.
Raw data file
The other file contains the raw data which enumerates the counts for each nucleotide at each position of interest. It also lists the occurrence of indels across the positions of interest and shows the number of times a sequence read contained a deletion, an insertion or neither (correct length). This file is tab-delimited text, best viewed in a spreadsheet program such as Excel. An example of this type of output file can be found here.
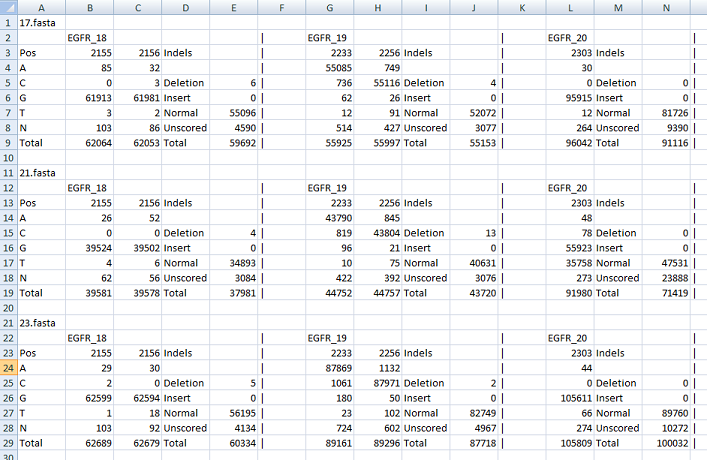
Figure 4: The raw data file format.