User guide
Introduction
AgileSMAll identifies rare somatic variants in clonal sequence data derived from a mixed population of cell types has found in tumour samples. The regions of interest must first be amplified by PCR and then clonally sequenced across the positions to be analysed. Unlike AgileSMPoint, this program does not look at specific positions but instead looks at all positions present in the sequence data. Since the sequence data, even when using multiplexed PCR products is derived from a relatively small population of read sequences, AgileSMAll is able to identify the origin of each read without prior alignment to target sequence. If the PCR products are longer than a typical read length it is possible to perform a paired ends sequence run and then combine the read data in to a single full length read using AgilePairedEndReadsCombiner.
Importing data
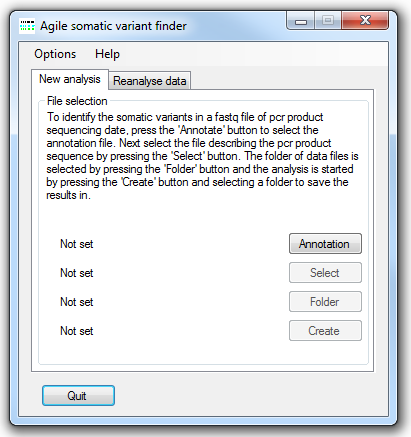
Figure 1: AgileSMAll user interface
AgileSMAll needs to import the sequence read data, a description of the genes in the genome and a description of the PCR amplicons in the experiment. To import a file describing the genome, press the button and select a genome annotation file. The creation of this file is described below in the Creating a genome annotation file section. Since this file is large it may take a moment to load. Once loaded the button will become active, press this button and select the file that describes the PCR products in the experiment. The format of this file is shown below in the PCR target file format section. Next select the folder of *.fastq files that you wish to analyse by pressing the button. Finally, to start the analysis press the button and select the folder you wish to save the somatic variant data in. A set of results files are created for each fastq file in the analysis. When AgileSMAll is processing a file, its name appears to the left of the button. When the analysis is complete a message box is displayed stating the analysis as finnished.
Creating a genome annotation file
AgileSMAll makes use of an annotation file that contains the locations of protein-coding exons and their genomic sequences and allows variants to be correctly annotated. This file can be created other programs in the Agile suite such as AgileAnnotator or AgileGenotyper. To create an annotation file, select the option on the menu (Figure 2). This causes the Annotation file creation window to be displayed (Figure 2).
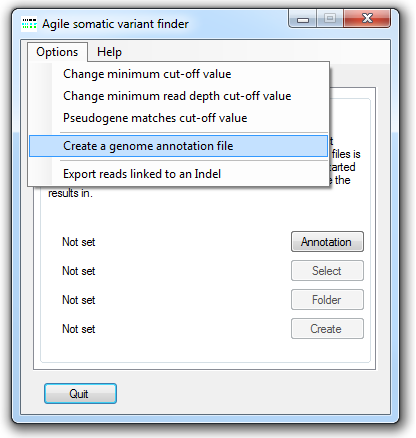
Figure 2: The annotation creation window
Note: The genomic sequences in the annotation files are derived from the current genomic sequence reference files and MUST match the version used when aligning the sequence reads to the genome and they MUST also be the same build version used by the creators of the CCDS or the RefSeq data files, used to identify the positions of coding sequences. If different reference sequences are used the analysis will fail!
The location of the uncompressed FASTA-format genomic reference files is selected using the button under Chromosome sequence files. These reference sequence files must follow the specific naming convention where each file name starts with "chr" followed by either the chromosome number or "X" or "Y", and has the .fa file extension. Permitted names include chr1.fa, chr5.fa for an autosome, while the X and Y may be named chrX.fa or chr23.fa and chrY.fa or chr24.fa, respectively.
Next select the source of the gene annotation file (CCDS or RefSeq) and then press the button in the CCDS data files panel and select the file containing the positions of the coding sequences as described by the Consensus CDS (CCDS) project or RefSeq datasets. The CCDS file can be downloaded from the NCBI CCDS web page or FTP site. While the RefSeq data file can be obtained from the UCSC Genome Browser's Tables page as described here.
Finally, press the button under Create annotation file and enter a name for the genomic annotation file. Since AgileSMAll has to read all of the sequences in the genomic reference files and then write a large amount of data, the creation of the annotation file may take several minutes.
Anaylsis options
Read depth based filtering
While the error rate for the calling of a specific position in a read is generally very low, due to the large number of positions analysed, the detection
of false variants can become problematic. Consequently, it is possible to adjust the minimum read depth and minimum cut off values at which a variant is
called (Figure 2). The option sets the minimum number of reads suggesting an alternative base as a
percentage of the total number of reads mapped to the base, i.e. ('number of variant bases' * 100) / ('number of variant bases' + 'number of reference bases').
The option sets the absolute minimum number of reads needed to suggest the presence of
an alternative base. Generally, speak the option is more important when there is a very high read
depth, while the is more important when the read depth is relatively low. These values
can be adjusted via the sub-menus in the menu.
Excluding reads derived from pseudogenes
The presences of highly homologous pseudogenes in the genome may lead to a proportion of sequence reads been derived from a pseudogene and not the functional gene. The presence of these reads
may either mask the true level of a mutant variant in the gene of interest or introduce false positives at positions where the pseudogene's sequence diverges from the gene's sequence. When
entering the amplicon's sequence in the target file (see below) positions in the lower case indicate that the sequence diverges between the gene and a pseudogene. The number of positions in a
sequence read that do not match the lower case bases is noted and if it is greater than the value set using the the read is discarded.
This value can be overridden for each target by entering an optional value in the PCR target file as described in the next section. This cut off value should not necessarily equal the number of
divergent positions especially if there is more than one pseudogene or the length of the amplicon is greater than the read length. Similarly it should not be set to 1 if gene conversion is important
or mutations at a divergent position are known to be important.
Exporting reads linked to indels
When scanning a read for possible indels AgileSMAll retains the first 1000 reads that suggest the presence of the same indel. If the
is checked, these reads will be exported to a text file saved in the same location as the other exported files. Reads complementary to
the forward are scanned for indels separately independently of reads complementary to the reverse strand. Consequently, two files may be created for each indel with the filename for the forward reads containing the
text '(+)' and the reverse reads containing the '(-)' text.
PCR target file format
The file used to describe the amplicons is structured in fasta file like format, the text description is place on a single line followed by the sequence data on the next line. This is then repeated for
each amplicon, with AgileSMAll able to scan many amplicons per experiment (Table 1).
Amplicon description
The information on the description line is separated by tab characters
and always starts with a '>' symbol followed by the name of the gene, a tab character and then the genomic coordinates of the first base of the amplicon sequence. This is the 5 prime most base of the amplicon
as it appears in the genome reference file. The last field is optional and should contain a positive number; for this target sequence, this value will override the
option previously described above in the Analysis Options section.
Sequence data
The data on the next line is also separated by tab characters with the first field containing the entire sequence of the PCR product including both primers. The sequence should be in
upper case characters except positions that diverge between the gene's sequence and a homologus pseudogenes sequence. See the earlier section Excluding reads derived from pseudogenes for a description of
how pseudogene sequence are detected. It is very important to note that the amplicons sequences should match the sequence of the + strand
in the reference sequence used to make the genome annotation file. This sequence is followed by the sequence of the forward and then the reverse primers. The sequence for the forward prime should match the sequence of
the amplicon as written in this file, while the reverse primer should be the reverse compliment of the amplicons sequence. An example of the files format can be found here. It is
recommended that you view this file as if this file is wrong, the analysis may appear to work, but will be wrong.
| >APC | chr5:112090560 | 1 (optional value) |
| PCR product sequence including primers | Forward primer | Reverse primer |
| >PIK3CA | chr3:178936048 | (optional value) |
| PCR product sequence including primers | Forward primer | Reverse primer |
| >PIK3CA | chr3:178952024 | 2 (optional value) |
| PCR product sequence including primers | Forward primer | Reverse primer |
| >PTEN | chr10:89692840 | (optional value) |
| PCR product sequence including primers | Forward primer | Reverse primer |
Table 1: The structure of file describing the sequenced PCR products.
Note the sequence of the primers is included in the PCR product's sequence.
Exported data file formats
The exported data is saved to three files per sample; the report file (Figure 3), the indel alignment file (Figure 4) and the raw data file (Figure 5) each file is described below. If the option is selected files containing upto 1000 forward or reverse reads will also be saved.
Variant report file
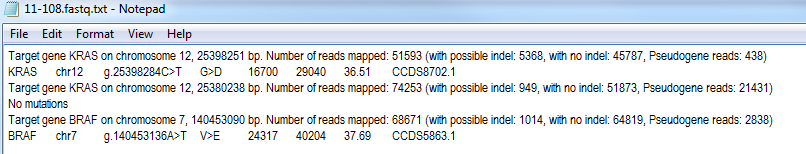
Figure 3: The somatic mutation report file format.
The report file is composed off a number of sections, with one section per PCR product listed in the target file. The first line of the section identifies the target gene and the location of the first base of the PCR product in the genome. This is followed by the number of reads mapped to the PCR product, with this value is broken down into the number of reads that may or may not contain an Indelfollowed by the number of reads identified as originating from a pseudogenes. Only reads without a possible indel are used to detect single base variants. The variants are then listed after this information. If no variants were detected in a PCR target then the phrase 'No mutations' is written otherwise the variants are listed one per line. For each variant the name the gene and its chromosome is shown, followed by the annotated variant and a description of the variants affect on the protein sequence. Typically this shows either the amino acid substitution or 'WT' if there is no change. Values of SP, In, KS and FS identify variants in a splice site, an intron, near the start codon or frame shift mutations respectively. This is followed by the variant allele's read depth, the read depth of the reference allele and the percentage of reads mapped to the variant allele. Finally, the name(s) of the affected transcript are listed.
Since the DNA is amplified by PCR prior to the sequencing it is possible for a number of PCR artifacts to be present in the read data which could be mistaken for reads contain an indel. In an attempt to identify these aberrant reads, the first six base after the end of a prime are compared to the expected sequence. If they differ the read is rejected, this results in fewer false indel variants, but also means that indels with in the six bases of a primer will not be detected. AgileSMAll attempts to independantly identify an indel in the forward and reverse reads. If the same indel is found in both directions the data for the forward and reverse reads are combined. If AgileSMAll can not find an indel in both directions, the indels are exported with and labelled with either a '(+)' or '(-)' to indicate which strand the reads originate from. If an indel is found in only one direction, and so has half the expected read depth, the indel is exported if it as more than half the minimum cut off read depth values, i.e. if a variant is exported when it is in more then 5% of reads, an indel found in only one direction will be exported if it is found in more than 2.5% of reads. If an indels genotype can not be correctly deduced the variant is shown as '>Indel'.
Indel alignment file
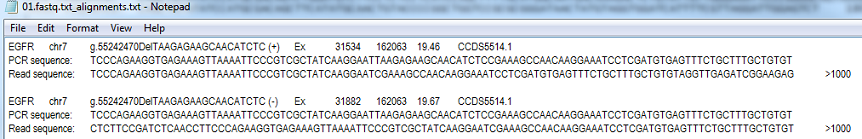
Figure 4: The indel alignment file format.
Reads that appear to contain an indel are scanned for the indel's start point and then sorted in a collection of reads that all appear to contain indels at the same location, with only the first 1000 reads are used to annotate the indel. These reads are combined in to a single sequence such that a position is called if more than 50% of reads contain the same base at that position, otherwise it is reported as an 'N'. This composite read is then compared to the amplicon's sequence to identify the insertion/deletion's genotype before been annotated and identical indels from both forward and reverse collections of reads combined. Even though the reads are first filtered for the presence of PCR artifacts a number of unannotable indels may be found. To allow the user to check these variants, the alignment used to identify a variant is saved to the indel alignment file after the indels description which is in the same format as in in the report file. At the end of the 'Read sequence' the number of reads used to create the alignment is shown, this value is never greater than 1000. AgileSMAll limits the maximum length and location of an indel such that the indel most start at least 6 bp after the primer sequence in the read and end more than 20 bp from the end of the sequence read.
Raw data file
The raw data file enumerates the read depth for each base at each position in the PCR product which is not part of a primer used to amplify the PCR product. By analysing this file it should be possible to check the error rate for each product. It should be noted that this data does not include data from reads that appeared to contain an indel.
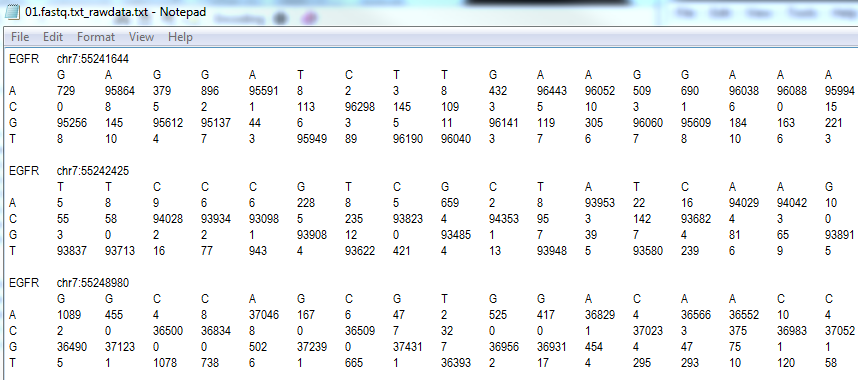
Figure 5: The raw data file format.
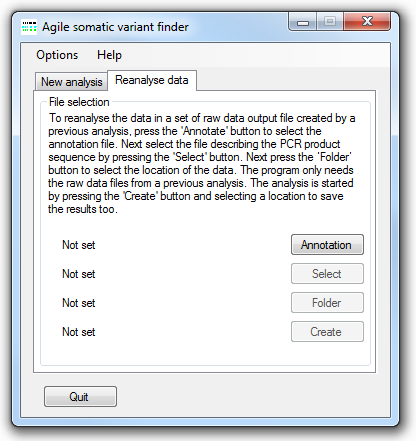
Figure 6: Reanalysing raw data files.
Since this data contains all the information used to detect single base somatic sequence variants, it is possible to reanalyse them using different cut off values. However, since this file does not contain information on reads containing possible indels it is not possible to rescreen of insertions and deletion. To reanalyse the data, select the Reanalyse data tab (Figure 6) and select the annotation and target files as before. Rather than selecting a folder of *.Fastq files, select the folded of raw data text files and then press the to start the analysis. It should be noted that for AgileSMAll to identify the correct data files the end of the file name must end with the 'rawdata.txt' text. The exported variant data is saved to a file with "_Reanalysed" appended to the original file name.