User guide
Introduction
AgilePairedEndReadsCombiner reads a matching pair of fasta or fastq files created from a paired end sequence run which used PCR products as the library template and combines the data to produce a single read file contain data equivalent to the whole PCR product. When comparatively short PCR products are sequenced to produce paired end reads the entire length of the PCR amplicon may be sequenced, but data is present in two different file. AgilePairedEndReadsCombiner is able to read matched paired sequence data files and identify the region of overlap between two reads from the same molecule and then combine them to form a single read. If the data is in fastq files, then the quality strings are also combined. If a position present in both reads and is different in each read, the position is described as an 'N' in the exported read. For positions common to both reads the highest quality score is retained in the exported quality string.
Selecting data from a single pair of data files
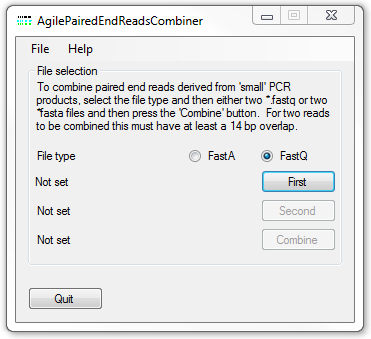
Figure 1: AgilePairedEndReadsCombiner user interface
To combine the data from a single pair of data files, first select the data file's format using the or options. The select the each file in turn using the or buttons. The order in which the files are added is not important. Finally press the button and enter the name of the file to save the data too. AgilePairedEndReadsCombiner will the read each pair of sequence reads in turn and where possible combining the reads.
Combining data from a set of single pair of data files
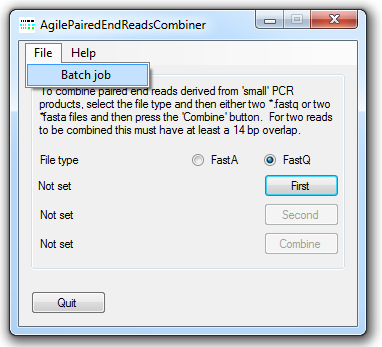
Figure 2: The menu allows data to be processed as a batch job.
In situations where there are a large number of data files to combine it is possible to run AgilePairedEndReadsCombiner in batch mode. To do this first create a text file that contains each pair of file names on a single row, separated by a tab character. An example of the format is shown in Table 1, while a file can be found here. It is important to note that you should only include the file names and not the entire file path. Place this file in the same folder as the data files and then select the file using the menu in the menu (Figure 2). AgilePairedEndReadsCombiner will use a file's extension to determine its format and then combine the sequence data in each file to a new file. This file will be saved to the same folder as the original data files and would be named by concatenating the names of the source files. The names of the current source files will be displayed to the left of the and buttons and the files are processed.
| A file in the pair | The other file in the pair | |
|---|---|---|
| 1_1.fastq | <tab character> | 1_2.fastq |
| 2_1.fastq | <tab character> | 2_2.fastq |
| 3_1.fastq | <tab character> | 3_2.fastq |
| 4_1.fastq | <tab character> | 4_2.fastq |
Table 1: The the structure of a 'batch job' file.
Note the file's path is not included with the file's name, but the file must be placed in the same data
folder as the data files.
Options for AgilePairedEndReadsCombiner
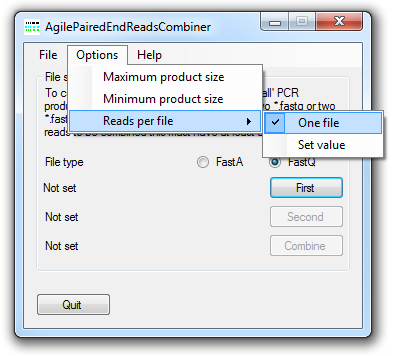
Figure 3: The menu.
Setting the size range of the PCR products.
The minimum and maximum size range of the PCR products in the dataset can be set using the and menus options (Figure 3). The possible size range can be from 40 to 1,000 bps.
Setting the maximum number of reads per file
It is possible to either save all the combined reads to a single file or a series of files. To save the reads to a single file select the option (Figure 3). To save the reads as a series of files select the option (Figure 3) and enter the maximum number of combined reads to save to a file. The data is saved to a series of files, each with as the same name except for an incremented integer value appended to it's name.