User guide
Introduction
AgileExomeFilter enables the filtering, screening and sorting of variants derived from an exome sequencing experiment to allow the rapid detection of possible deleterious variants. The variants must first be annotated using Alamut-HT and users may find it useful to read about Alamut-HT before continuing.
Entering data
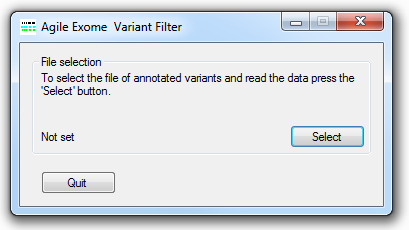
Figure 1: AgileExomeFilter file selection user interface
The Alamut-HT annotated variant data file is imported by pressing the button (Figure 1) and selecting the required text data file.
The variant display interface
AgileExomeFilter displays the variant data as a grid of 40 columns containing the first 100 variants in the data set. If the filtered dataset includes more than 100 variants it is possible to navigate through them as a series of pages (100 variants per page), using the value in the Filter variants panel in the bottom right of the window (Figure 2).
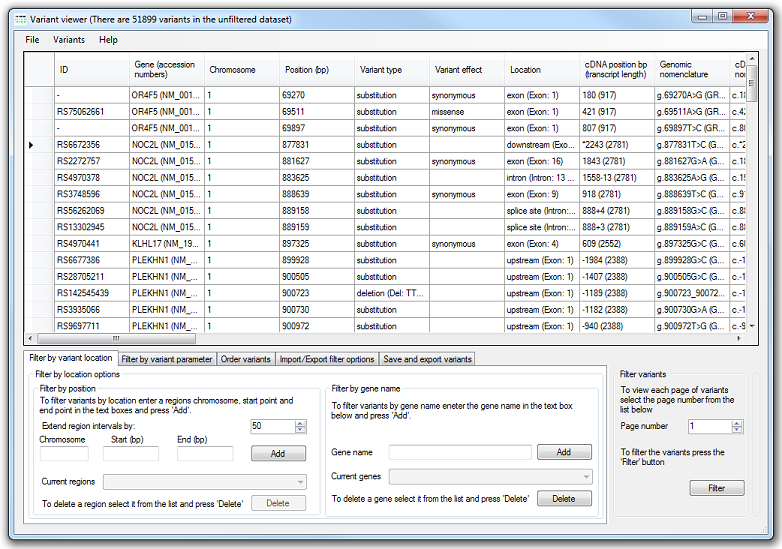
Figure 2: The variants are displayed as a grid.
The data for each variant are shown on a single row, with the columns containing the data from the Alamut-HT data file. To make the data easier to view some of the Alamut-HT fields are combined into a single column, while others containing duplicated data are ignored. The variant data view is described in Table 1 below:
| Column Name | Description | Example or possible values | Link to more information |
|---|---|---|---|
| ID | The variant’s RS number | RS6672356 | dbSNP |
| Gene (accession numbers) | Name of the gene plus the transcript and protein accession number | NOC2L (NM_015658.3 | NP_056473.2) | HGNC and RefSeq |
| Chromosome | The chromosome containing the variant | 1-22,X,Y | |
| Position (bp) | Location of the variant in base pairs | 877831 | |
| Variant type | Type of mutation | deletion, duplication, insertion or substitution | |
| Variant effect | Effect of the variant | frameshift, in-frame, missense, synonymous | |
| Location | Location of the variant within the gene | downstream (Exon: 19) | |
| cDNA position bp (transcript length) | Location of the variant within the transcript/cDNA and its total length | 2243 (2781) | |
| Genomic nomenclature | Variant annotated relative to genome coordinates | g.877831T>C (GRCh37) | HGVS |
| cDNA nomenclature | Variant annotated relative to transcript/cDNA coordinates | c.2243A>G | HGVS |
| Codon change | Reference and variant codon | TCT>TCC | |
| Protein nomenclature | Description of variant in the protein sequence | p.Ser269Ser, p.Phe1182* | HGVS |
| OMIM ID | Omim reference ID of the gene containing variant | 610770 | OMIM |
| SSF splice site ratio (var/wt) | The ratio of the SSF splice site score for the nearest splice site with the variant sequence divided by the value of the reference sequence. Should be 1 for no effect. | -infinity to infinity | SSF |
| MaxEnt splice site ratio (var/wt) | The ratio of MaxEnt splice site score for the nearest splice site with the variant sequence divided by the value of the reference sequence. Should be 1 for no effect. | -infinity to infinity | MaxEntScan for human 5' sites |
| NNS splice site ratio (var/wt) | The ratio of NNS splice site score for the nearest splice site with the variant sequence divided by the value of the reference sequence. Should be 1 for no effect. | -infinity to infinity | |
| GS splice site ratio (var/wt) | The ratio of GS splice site score for the nearest splice site with the variant sequence divided by the value of the reference sequence. Should be 1 for no effect. | -infinity to infinity | |
| HSF splice site ratio (var/wt) | The ratio of HSF splice site score for the nearest splice site with the variant sequence divided by the value of the reference sequence. Should be 1 for no effect. | -infinity to infinity | HSF |
| Nearest splice site change | Location of nearest splice site to the variant. | ||
| Splicing effect in vicinity | Describes the possibility of the variant creating a splice site | cryptic acceptor strongly activated, cryptic donor strongly activated, new acceptor site, new donor site | |
| RS is validated | If the variant has a RS number, has the variant been validated | yes or no | dbSNP |
| Clinical Significance | The variant's clinical significance as stated by dbSNP | unknown, untested, non-pathogenic, probable-non-pathogenic, probable-pathogenic, pathogenic, drug-response, histocompatibility, other | dbSNP |
| Minor allele dbSNP frequency | Minor allele frequency if variant is in dbSNP (List of alleles in different populations) | e.g. 0.005 (C,C,C) | dbSNP |
| Minor allele dbSNP count | Number of times alleles found in dbSNP data | e.g. 2188 | dbSNP |
| African American allele data (ESP) | Allele frequency in African American population | 0 to 1 | NHLBI Exome Sequencing Project |
| European American allele data (ESP) | Allele frequency in European American population | 0 to 1 | NHLBI Exome Sequencing Project |
| All population allele data (ESP) | Allele frequency in all populations | 0 to 1 | NHLBI Exome Sequencing Project |
| Average read depth (ESP) | Average read depth of data used to identify SNP in ESP data set | e.g. 122 | NHLBI Exome Sequencing Project |
| PhastCons Score | PhastCons severity score of variant | e.g. 1 | PhastCons |
| PhyloP score | PhyloP score severity score of variant | e.g. 1.981 | PhastCons |
| BLOSUM 45, 62 and 80 scores | BLOSUM 45, 62 and 80 matrix severity score of the variant based on the evolutionary rate of amino acid substitution | i.e. -3,-3,-6 | Wikipedia |
| SIFT prediction score | SIFT severity score for the variant | deleterious, tolerated | SIFT |
| SIFT weight | Variant's SIFT weight score | 0 to 1 | SIFT |
| SIFT median | SIFT scores median value | SIFT | |
| Grantham distance | The severity of the variant based on the Grantham distance | > 0 | Grantham distance |
| VCF quality score | The VCF quality score for the variant | > 0 | |
| Genotype | Genotype of the variant | homozygous, heterozygous | |
| Total read depth | Number of reads mapping to position | > 0 | |
| Allele read depths | The number of reads containing the variant and reference base | >0, >0 | |
| Pathogenicity class | User-defined classification of the variant if previously annotated. Value must start with 'Class ' and the a value of 1 to 5. | Class 1, Class 2, Class 3, Class 4, Class 5 |
Table 1: Description of the columns in the data grid.
Filtering variants based on their genomic location
It is possible to filter the variants based on their position in the genome and/or location in a gene.
If variants are filtered by both regions and genes, a variant is retained if it is present in either a region or a gene.
Filter by position
To filter a set of variants based on their location, enter a region in the
Filter by position panel on the Filter by variant location tab at the bottom of the window (Figure 3). This is done by entering
the region's chromosome, start and end positions in the text boxes and pressing . The sex chromosomes are entered as either X or Y, while the
mitochondrial chromosome is entered as an M. To delete a region, select it from the list and press .
It is important that these regions should not overlap.
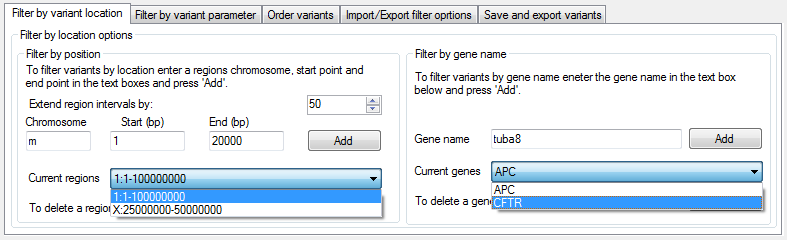
Figure 3: The Filter by variant location tab
It is also possible to import regions saved in a text file with the format shown in Table 2, using > . For example, using such a file it is possible to import data relating to all the exons in a pull-down experiment. Since the exon regions specified in such a file may not include flanking regions that may also be of interest, it is possible to extend the regions into the flanking sequence by up to 50 bp, using the Extend region intervals by: option (located above the button). Regions imported from a file do not appear in the list and so cannot be deleted; to delete them first close the current window and then reenter the variant data file.
| Chromosome | Separator | Start point (bp) | Separator | End point (bp) |
|---|---|---|---|---|
| chr1 | : | 105000000 | - | 153500000 |
| chr4 | : | 5000000 | - | 6500000 |
| chrX | : | 85000000 | - | 93500000 |
Table 2: The format of a file used to import region data.
Filter by gene
To filter the variants by the name of the gene within which they are located, enter the gene name in the Gene name text box in the Filter by gene name panel on the Filter by variant location tab at the bottom of the window (Figure 3). To delete a gene name, select it from the list and press the button. It is also possible to enter a list of gene names using the > and selecting a text file in which each line contains a single gene's name.
Filter by variant characteristic
It is possible to filter variants against a number of different parameters, using the options in the Filter by variant properties options panel on the Filter by variant parameter tab at the bottom of the window (Figure 4).
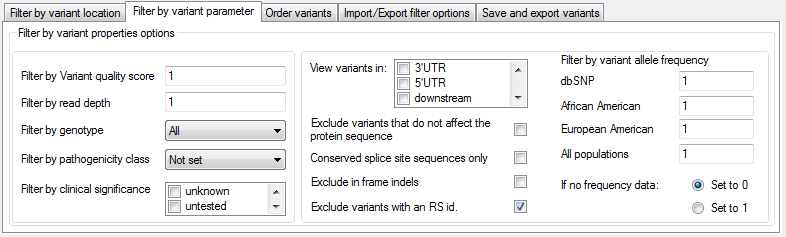
Figure 4: The Filter by variant parameter tab allows variants to be filtered against a number of different characteristics.
Each option is described below:
- Filter by variant quality score: filters the variants by the VCF quality score calculated by the variant-calling program.
- Filter by read depth: filters the variants by the total read depth at the variant's position.
- Filter by genotype: allows the selection of all variants or only those that are either homozygous or heterozygous. It is also possible to select for variants in genes that contain at least two heterozygous variants, using the Biallelic option (homozygous variants will also be retained).
- Filter by pathogenicity class: filters the variants by the user-defined pathogenicity class which is used to annotate variants previously described in the user's group or institute.
- Filter by clinical significance: filters the variants based on their pathogenicity as described by dbSNP.
- View variants in: filters variants based on their location within a gene.
- Exclude variants that do not affect the protein sequence: filters the variants located in exons based on their effect on the protein sequence.
- Conserved splice site sequences only: retains variants within two base pairs either side of a splice site.
- Exclude inframe indels: discords indels in exons that do not cause a frameshift in the protein sequence.
- Exclude variants with an RS id: excludes variants with an RS number.
- Filter by variant allele frequency: filters the variants based on the allele frequency of the minor allele as described by either dbSNP or ESP. The maximum minor allele frequency is set using the appropriate text box and should be a number from 0 to 1.
- If no frequency data: option sets a variants minor allele frequency to either 0 or 1 if no allele frequency data is present in the Alamut-HT file for that variant.
Filtering the data
Once the parameters have been selected, the variant data can be filtered by pressing the button on the Filter variants panel in the bottom-right corner of the window (Figure 2). The number of variants remaining after filtering will be displayed in the window's title bar (Figure 5).
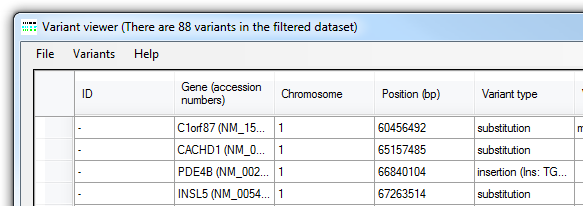
Figure 5: The number of variants not excluded by the filtering process is shown in the title bar.
Sorting variants
The Sort variants by panel on the Order variants tab (Figure 6) allows variants to be sorted based on 12 different parameters, in either ascending or descending order. The list contains the fields by which the variants may be sorted, while the and options set the sorting order.
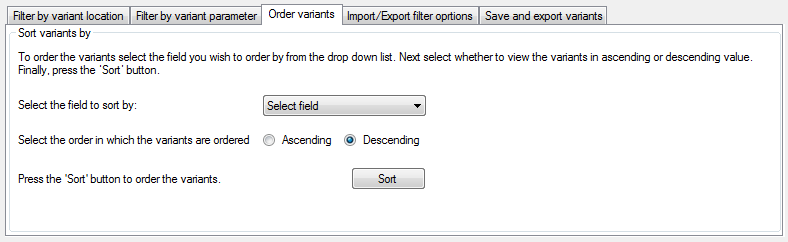
Figure 6: The Order variants tab allows variants to be sorted on different parameters.
Importing and exporting filtering parameters
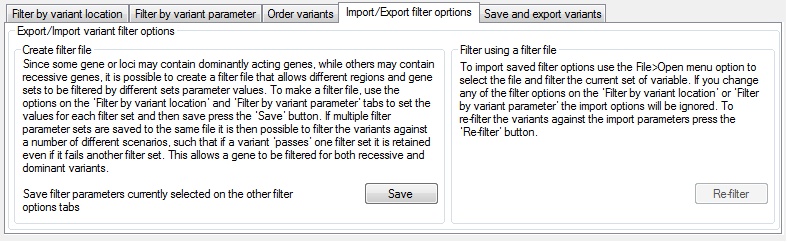
Figure 7: The Import/Export filter options tab allows filter options to be reused.
The Import/Export filter options tab (Figure 7) describes how to save a set of variant filter options, so that the same parameters can be rapidly and consistently used to screen multiple datasets. Once the filtering options have been set, it is possible to save them by pressing the button. This appends the filter options to the selected file, allowing multiple filter criteria sets to be present in one file. If multiple filter criteria sets are imported from a single file, the variants are filtered against each set of criteria in turn, with variants retained if they past at least one of the filter sets. This allows a clinical disorder such as deafness (which may be dominantly or recessively inherited) to be screened using multiple filtering sets; one set of parameters suited for variants in genes with a recessive mode of inheritance, while the second set of criteria is tailored for variants in genes with dominant inheritance pattern.
Filter options saved to a text file are imported using > . When imported, these filter options remain distinct from any parameters specified using the user interface, which will be ignored during this filtering process. Conversely, if the button described in the Filtering the data section is used, the imported filter parameters are ignored. To refilter the variants using the imported values, press the button in the Filter using a filter file panel (Figure 7).
Saving and exporting variants
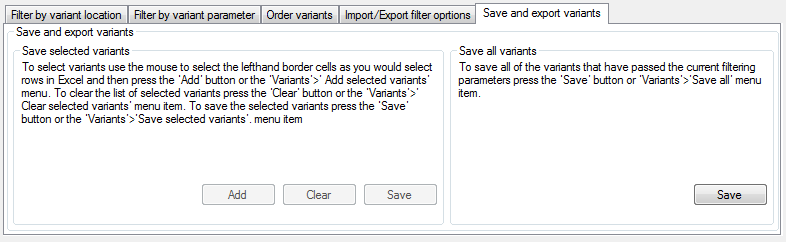
Figure 8: The Save and export variants tab allows filter options to be reused.
It is possible to save either all variants that have passed the filtering process, or a smaller user-selected set of variants. This can be done either via the menu or using the Save and export variants tab (Figure 8). To save all variants, either press the button in the Save all variants panel or select the > menu option and enter the name of the target file. To save a user-selected list of variants, select the variants using the data grid in the main window (in the same way that you would select data rows in Excel) and then either press the button in the Save selected variants panel or select the > menu option. This process can be repeated multiple times to create a collection of variants. Once the variants have been added, it is possible to refilter the data using a different set of parameters and then add more variants to the saved variants. The saved variants are the exported to a text file by either pressing in the Save selected variants panel or selecting the > menu option. Once the variants have been saved, the user will be asked if the stored variants should be cleared. To clear the stored variants without first saving them, either press in the Save selected variants panel, or select the > menu option.